
Влияние умеренной гипогликемии на уровень белка S100b в крови и гиппокампе крыс
Мазукина Е.В., Шекунова Е.В., Кашкин В.А., Фаустова Н.М., Макаров В.Г. Влияние умеренной гипогликемии на уровень белка S100b в крови и гиппокампе крыс. Лабораторные животные для научных исследований. 2019; 1. https://doi.org/10.29296/2618723X-2019-01-07
Резюме
Значительное снижение уровня глюкозы в крови ведет к развитию отсроченных нарушений когнитивных функций, причиной которых могут быть вызванные гипогликемией повреждения нейронов головного мозга. В экспериментах на крысах показано, что как умеренная, так выраженная гипогликемия, вызванная введением инсулина, приводит к повреждениям нейронов в нескольких областях головного мозга, включая неокортекс и гиппокамп. В проведенном исследовании умеренную гипогликемию индуцировали ежедневным подкожным введением инсулина аспарта (15 ЕД/кг в течение 5 дней), которую через 60 мин купировали внутрибрюшинным введением 25% раствора глюкозы. Когнитивный дефицит оценивали в тесте «Водный лабиринт Морриса» на 35-й день эксперимента. Учитывая наибольшую подверженность нейронов гиппокампа негативному действию гипогликемии, для оценки повреждений нервной ткани был проведен иммуногистохимический анализ экспрессии белка S100b в области СА1 гиппокампа, параллельно была изучена динамика изменения концентрации S100b в плазме крови. Введение инсулина приводило к развитию эпизодов умеренной гипогликемии (концентрация глюкозы в крови – 2,1 ммоль/л) продолжительностью 60 мин. При оценке когнитивных функций в тесте Морриса было показано, что животные со сформированной патологией, в отличие от контрольных, не «патрулировали» сектор, в котором на этапе обучения находилась платформа. Продолжительность пребывания в данном секторе была меньше по сравнению с показателями в контрольной группе (t-тест; p<0,05). Оценка концентрации белка S100b в плазме крови показала, что непосредственно после формирования патологии и до 35 дня эксперимента у животных, перенесших эпизоды гипогликемии, уровень белка был существенно повышен по сравнению с контрольной группой. Увеличение концентрации белка в крови коррелировало с увеличением экспрессии белка S100b в зоне СА1 гиппокампа к 35-му дню эксперимента. Полученные результаты свидетельствует о том, что изменение концентрации S100b является важным диагностическим маркером развития повреждения головного мозга, который можно оценивать в динамике и использовать для изучения эффективности новых лекарственных препаратов.
Введение
Как известно, значительное снижение уровня глюкозы крови, главного источника энергии головного мозга, ведет к развитию отсроченных нарушений когнитивных функций, причиной которых могут быть вызванные гипогликемией повреждения нейронов [1, 2].
Среди основных причин развития повреждений головного мозга на фоне гипогликемического состояния выделяют оксидативный стресс, а также индукцию эксайтотоксических процессов [3]. Снижение интенсивности метаболизма глюкозы в нейронах ведет к снижению запасов энергии в форме АТФ и НАДH, с параллельным увеличением образования возбуждающих аминокислот, таких как аспартат и глутамат [4]. Баланс между тормозными и возбуждающими аминокислотами смещается в сторону последних, именно поэтому при значительном снижении уровня глюкозы крови могут наблюдаться судороги [1].
Увеличение образования возбуждающих аминокислот является причиной гибели нейронов. Однако гипогликемия индуцирует и другие биохимические изменения, многие из которых противоположны тем, которые наблюдаются при ишемических поражениях головного мозга [1]. Так, например, при гипо-
гликемии развивается тканевой алкалоз за счет увеличения синтеза аммиака в процессе катаболизма белков и дезаминирования аминокислот. Накопление аммиака приводит к увеличению рН клетки. Другой причиной развития алкалоза при гипогликемии является недостаток пирувата вследствие снижения интенсивности обмена глюкозы. Именно поэтому на фоне гипогликемии не развивается инфаркт ткани головного мозга, поскольку нет возможности снижения рН в клетках. Это, в сочетании с отсутствием ишемии, объясняет, почему на фоне гипогликемии наблюдается избирательный нейронный некроз («выпадение нейронов»), но не инфаркт [1].
У пациентов, которые перенесли тяжелую гипогликемию, наблюдается ухудшение когнитивных функций, особенно в отношении кратковременной памяти, при этом изменений общей двигательной активности не происходит [5]. Схожая картина отмечается при моделировании гипогликемического состояния у животных. У крыс после моделирования экспериментальной гипогликемии локомоторная активность не изменялась, однако формировался когнитивный дефицит – ухудшался процесс обучения и воспроизведения навыка при тестировании в тесте «Водный лабиринт Морриса» [6].
Экспериментальные исследования выявили, что более всего подвержены негативному влиянию гипогликемии нейроны гиппокампа и коры больших полушарий головного мозга [1, 6–10]. В экспериментах на крысах показано, что умеренная гипогликемия продолжительностью 60 мин, которая достигается ежедневным введением инсулина в течение 5 дней, вызывает повреждение нейронов в нескольких областях мозга, включая неокортекс, гиппокамп, структуры стриопаллидарной системы (скорлупа, хвостатое ядро) [11, 12]. Описанная схема индукции вызванных гипогликемией нарушений была использована в проведенном исследовании. Когнитивный дефицит оценивали при помощи теста «Водный лабиринт Морриса», который, согласно данным литературы, является информативным методом оценки нарушений процессов обучения и памяти [13, 14].
Известно, что одним из маркеров повреждения нервной ткани является нейроспецифичный белок S100b. В физиологических (наномолярных) концентрациях S100b защищает нейроны от апоптоза, стимулирует пролиферацию астроцитов, подавляет реакцию астроцитов и микроглии на нейротоксические воздействия. При повреждениях нервной ткани различной природы концентрации этого белка увеличиваются до микромолярных, поэтому значительное повышение его уровня рассматривается как признак поражения нейрональных структур [15]. Таким образом, белок S100b – неспецифический маркер повреждения головного мозга, реагирующий на механические, гипоксические, ишемические, биохимические и другие нейротоксические факторы. Выявлено, что повторяющиеся эпизоды умеренной гипогликемии (снижение уровня глюкозы до 2 ммоль/л) вызывают гибель нейронов, особенно в гиппокампе (СА1) [13, 16]. С учетом этого для оценки степени вызванных гипогликемией повреждений нервной ткани в данном исследовании был проведен иммуногистохимический (ИГХ) анализ степени экспрессии белка S100b в области СА1 гиппокампа, параллельно была изучена динамика изменения концентрации маркера в плазме крови после индукции патологии.
Материал и методы
Эксперименты выполнены на самцах аутбредных крыс массой 220–260 г (питомник НПО «Дом Фармации», Россия). Животных содержали в условиях 24-часового фоторежима (12 ч – день : 12 ч – ночь, включение света в 8:00), контролируемой температуры (22±2ºC) и влажности (65±10%) воздуха при свободном доступе к очищенной воде и стандартному корму (гранулированный комбикорм).
Данная научно-исследовательская работа была рассмотрена на биоэтической комиссии НПО «Дом Фармации» и одобрена для проведения (№БЭК 1.17/16 от 16.05.2016).
Дизайн исследования
В каждой экспериментальной группе было по 6 животных. Гипогликемию индуцировали подкожным введением инсулина животным 2-й группы, 1-я (контрольная) – 0,9% изотонический раствор NaCl подкожно. В остальном все манипуляции были аналогичны для 1-й и 2-й групп.
Для индукции гипогликемии НовоРапид Пенфилл® [инсулин аспарт, ООО «НовоНордикс», Россия (инсулин ультракороткого действия – начало эффекта через 10–20 мин после подкожного введения; пик 0,5–2 ч; длительность 3–5 ч) вводили ежедневно подкожно в дозе 15 ЕД/кг в течение 5 дней [11, 12]. Перед каждым введением инсулина экспериментальные животные были лишены корма на 8 ч. Состояние гипогликемии прекращали введением раствора глюкозы (25% раствор) (ЗАО «Биннофарм», Россия), внутрибрюшинно, 2 мл на животное, через 60 мин после каждого ежедневного введения инсулина. Перед введением глюкозы (через 60 мин после введения инсулина) и через 30 мин после введения глюкозы (90 мин после введения инсулина) измеряли концентрацию глюкозы в крови для контроля гипогликемического состояния.
Cохранность когнитивных функций (обучение и пространственная память) оценивали с помощью теста «Водный лабиринт Морриса» [14]. Обучение проводили на 29–32-й дни эксперимента, тест – на 34 й день после формирования патологии.
Взятие образцов крови для определения концентрации белка S100b выполняли из хвостовой вены на 6-й, 17-й и 35-й дни после начала индукции патологии. Взятие образцов материала для последующего ИГХ исследования проводили на 35-й день эксперимента (n=3 для каждой группы).
Определение уровня глюкозы в периферической крови
Для определения глюкозы в крови использовали глюкометр Accu-Chek® и тест–полоски Accu-Chek® Active. Животное помещали в «рестрейнер», проводили обработку хвоста антисептиком, делали прокол хвостовой вены и выделяющуюся каплю крови наносили на тест-полоску глюкометра.
Определение белка S100b в плазме крови
Кровь отбирали (0,7 мл) из хвостовой вены – на 6-й день эксперимента (1 «точка», исходный уровень сформированного патологического состояния); 17-й и 35-й день эксперимента (2-я и 3-я временные точки).
Подготовка проб для определения биохимических показателей
Кровь отбирали в пробирки типа Эппендорф с 7 мкл раствора гепарина натрия концентрацией 5000 МЕ/мл, которые далее центрифугировали (1000 g в течение 20 мин) и отделяли плазму (супернатант).
Концентрацию белка S100b в плазме крови животных определяли с помощью коммерчески доступных ИФА-наборов Elisa kit For S100 Calcium Binding Protein B (S100B) (Rat) (№ SEA567Ra, Cloud-Clone Corp., США). Линейность методики – 3,12 – 400 пг/мл.
Оценка когнитивных функций
Сохранность когнитивных функций оценивали с помощью теста «Водный лабиринт Морриса». Экспериментальная установка представляла собой круглый бассейн (d=150 см, h=60 см), наполненный до отметки
30 см. В одной из четвертей бассейна (в период обучения) находилась платформа, на 2 см ниже уровня воды. Бассейн был условно разделен на 4 равных сектора, которые обозначались буквами E, S, N, W.
Обучение проводили при искусственном красном освещении в период с 10 до 13 ч. Как показали результаты пилотных экспериментов, животные эффективнее обучаются в таких условиях. Во время обучения после обнаружения платформы или после того, как исследователь помогал ее находить, если животное не могло самостоятельно найти платформу в течение 60 с, крыса 15 с находилась на платформе для запоминания ее позиции.
Экспериментальных животных обучали с 29-го по 32-й дни эксперимента
(4 дня). Ежедневно животным предоставляли 4 попытки по 60 с. Платформа всегда находилась в позиции SW. Старт (посадку животных в бассейн) осуществляли с положений E, SE, N, NW, которые выбирались в случайном порядке. Тестировали животных на 34-й день эксперимента при красном освещении. Во время теста платформу убирали, старт теста был с новой позиции – NE.
Регистрируемые параметры:
Обучение:
- латентный период нахождения платформы на этапе обучения;
Тест:
- латентный период вхождения в сектор, где на этапе обучения была платформа;
- общее время, проведенное в секторе, где на этапе обучения была платформа;
- количество посещений сектора, где на этапе обучения была платформа;
- среднее время на 1 посещение в секторе, где на этапе обучения была платформа (общее время, проведенное в секторе, деленное на количество посещений сектора).
ИГХ исследование
Для получения материала с целью проведения ИГХ исследования животных наркотизировали внутримышечным введением смеси препаратов Zoletil 100 (2,6 мг/кг) и Xila (2,6 мг/кг), после чего проводили перфузию 10% нейтральным раствором формалина [17]. Затем головной мозг извлекали [18], фиксировали в 10% растворе нейтрального забуференного формалина в течение 24 ч. На этапе вырезки фронтальные срезы головного мозга крыс выполнялись на уровне 33–34, согласно атласу L.W. Swanson [19]. Затем материал был подвергнут стандартной гистологической обработке с этапами промывки в проточной воде, дегидратации в изопропаноле возрастающей концентрации и заливки в парафин [20, 21].
В работе были применены кроличьи моноклональные антитела к S-100 beta (клон 16H24L21, ABfinityTM, ThemoFisher) в разведении 1:75. В качестве систем визуализации для первичных антител были использованы вторичные поливалентные антитела Histofine Simple Stain MAX PO (multi) и хромоген Histofine DAB-2V (Nichierei Biosciences Inc.). Постановку ИГХ-реакций выполняли согласно протоколам, предоставленным производителями антител.
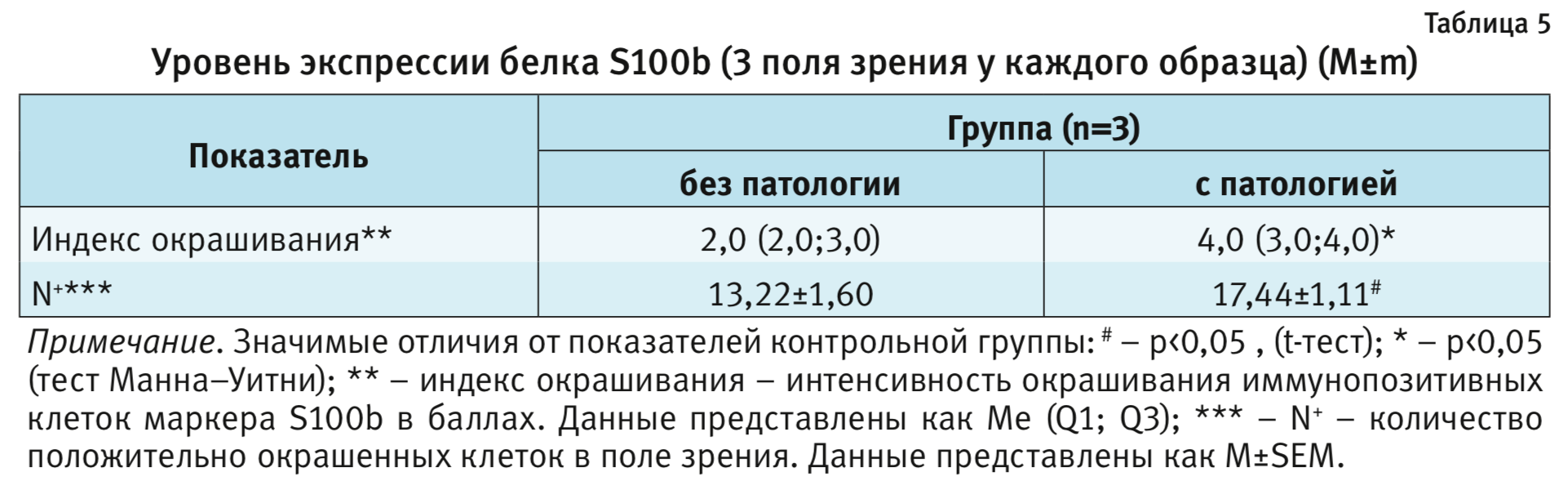
Морфологический анализ осуществляли при помощи светооптического микроскопа Carl Zeiss Axio Scope A1 (Германия) при увеличении 200; микрофотографирование – при помощи цифровой фотокамеры AxioCamICc 1 и программного обеспечения AxioVisionRel. 4.8 (Германия). Оценку экспрессии белка S100b проводили не менее чем в 3 полях зрения. Подсчитывали иммунопозитивные клетки, а также оценивали полуколичественную интенсивность ИГХ окрашивания (в баллах, от 0 до 5) согласно следующим критериям:
0 – отсутствие иммунопозитивных клеток в поле зрения;
1 – незначительная экспрессия маркера в иммунопозитивных клетках;
2 – большая часть клеток со слабой экспрессией маркера;
3 – умеренная экспрессия маркера в иммунопозитивных клетках;
4 – иммунопозитивные клетки с умеренной и выраженной экспрессией маркера;
5 – большая часть клеток с выраженной экспрессией маркера.
Статистическая обработка. Межгрупповые различия анализировали параметрическими или непараметрическими методами, в зависимости от типа распределения. Для оценки данных с признаками нормального распределения было использовано парное сравнение групп при помощи t-теста, в случае если распределение не подчинялось закону нормального распределения – тест Манна–Уитни. Для анализа данных использовался двухфакторный дисперсионный анализ (ANOVA) с повторными измерениями (факторы: группа и время), в случае обнаружения значимого влияния исследуемого фактора post hoc сравнения выполняли с помощью критерия Бонферрони. Для данных, не подчиняющихся закону нормального распределения, прибегали к тесту Манна–Уитни. Различия были определены при 0,05 уровне значимости. Статистический анализ выполнялся с помощью программного обеспечения Statistica 10.0. (StatSoft, США).
Результаты и обсуждение
Моделирование гипогликемии. Измерение уровня глюкозы.
Исходная масса тела экспериментальных животных составила 240,00±7,36 г. Измерение уровня глюкозы в крови проводили через 1 ч после введения инсулина. У всех животных через 1 ч после каждого ежедневного введения инсулина наблюдалась выраженная гипогликемия: концентрация глюкозы в крови была снижена по сравнении с контрольной группой (табл. 1, двухфакторный дисперсионный анализ ANOVA с повторными измерениями F1,10=176,0; р<0,001). Последующие межгрупповые сравнения подтвердили снижение концентрации глюкозы в периферической крови по сравнению с показателями в группе без патологии (р<0,05; критерий Бонферрони). Показатели находились в диапазоне от 1,3 до 2,6 ммоль/л. На фоне гипогликемии активность животных снижалась, при этом эпизодов судорог не наблюдали. Состояние гипогликемии купировали введением раствора глюкозы. Уровень глюкозы в крови в течение 90 мин после введения инсулина нормализовался и не отличался от таковой в группе контроля. Таким образом, у животных с индукцией патологии в течение 5 дней формировали состояние гипогликемии продолжительностью 60–70 мин.
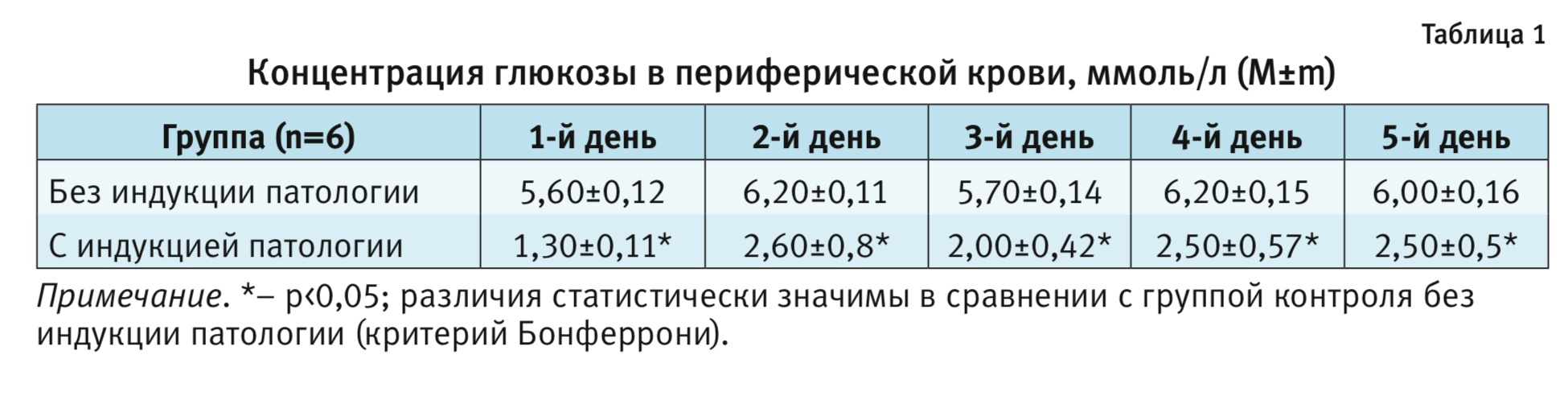
Поведенческое тестирование. Оценка когнитивного дефицита. Тест «водный лабиринт Морриса»
Тестирование было разделено на 2 этапа: 1-й – обучение, продолжительность 4 дня; 2-й этап – тест, в котором оценивали процесс консолидации полученной при обучении информации.
Все животные хорошо обучались, различий между группами с патологией и без патологии не обнаружено. При проведении статистического анализа данных с применением двухфакторного дисперсионного анализа (ANOVA) с повторными измерениями (1-й фактор – «группа», 2-й – «время») влияния фактора «группа» на латентный период нахождения платформы не выявлено (F1,10=0,4; p=0,54, табл. 2). Однако статистически значимым было влияние фактора «время» (F3,30=19,4; р<0,001). Последующее межгрупповое сравнение подтвердило укорочение латентного периода, начиная со 2-го дня (30–32 дни обучения) по сравнению с 1-м днем (р<0,05; критерий Бонферрони; см. табл. 2), что свидетельствует об эффективности обучения.
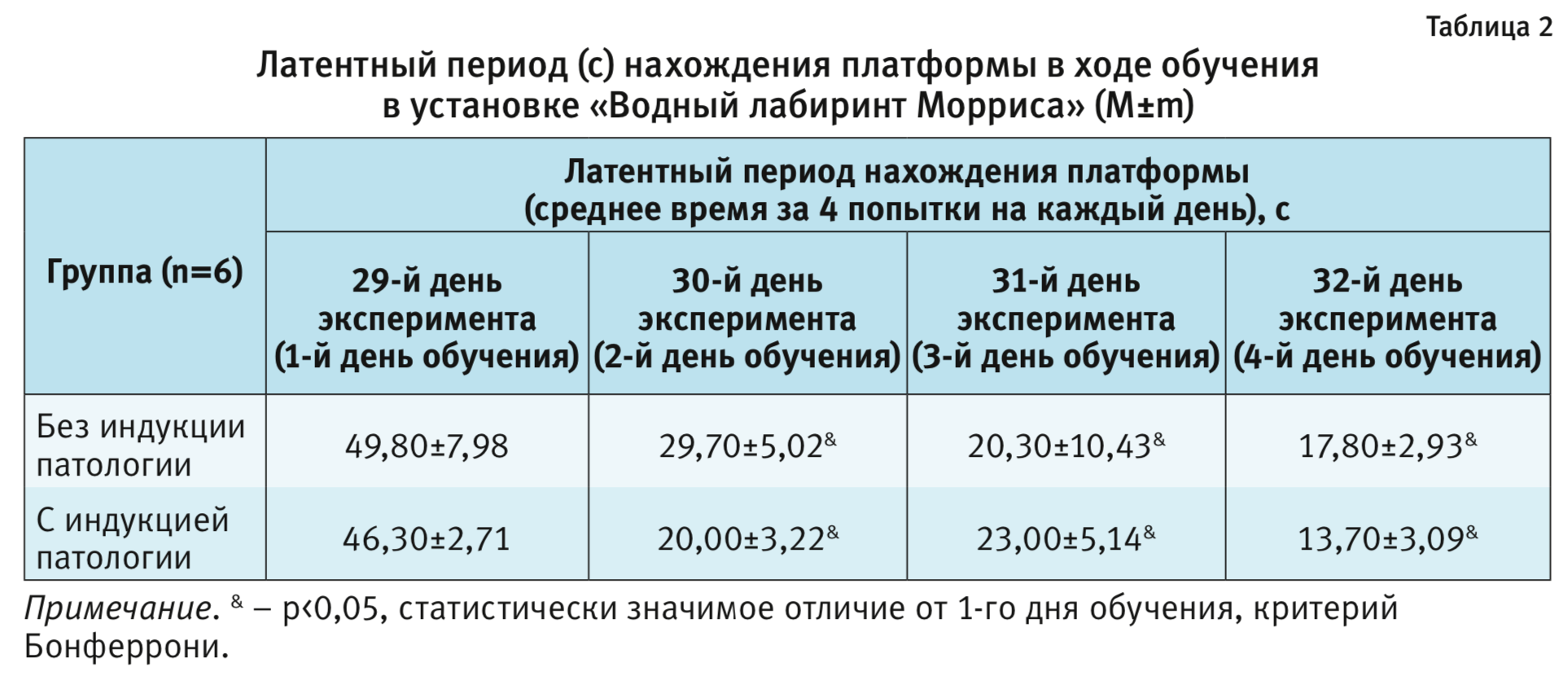
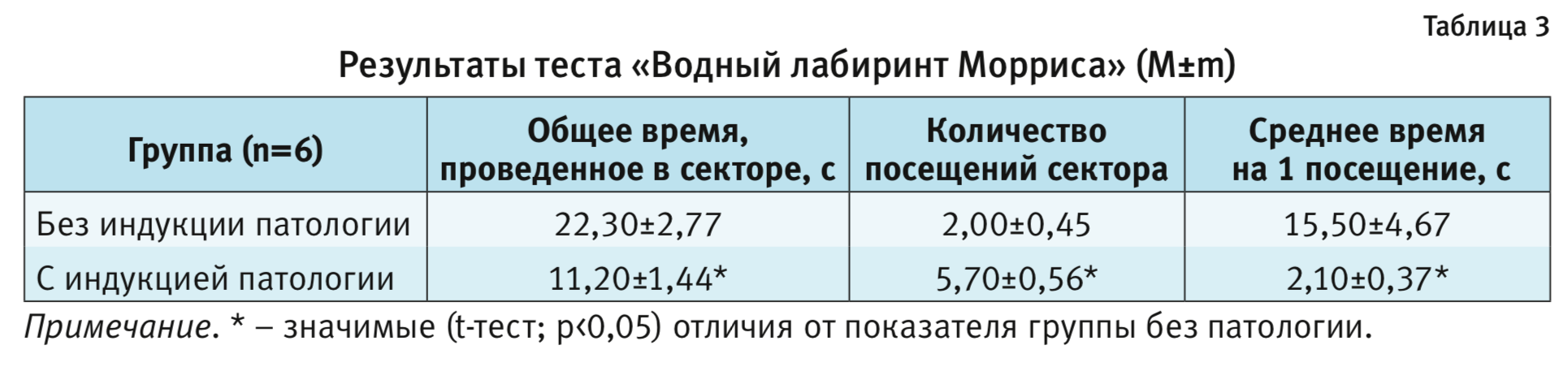
Через 48 ч после обучения животные были протестированы на воспроизведение полученного навыка. Проведенное парное сравнение не показало статистически значимого отличия по показателю – латентный период входа в сектор, где при обучении находилась платформа, между группой с патологией (13,6±2,3 с) и контрольными животными (13,8±3,3 с) (р>0,05, t-тест). Парное сравнение животных группы с патологией и контрольной группы выявило статистически значимые различия по показателям «общее время, проведенное в секторе», «количество посещений сектора» и «среднее время на 1 посещение» (р<0,05, t-тест, табл. 3). Следует отметить, что стратегия поведения животных со сформированной патологией отличалась от таковой у контрольных животных. Поведение первых было менее целенаправленным. Хотя они чаще, чем контрольные животные, посещали сектор, где в период обучения находилась платформа, но, в отличие от контрольных, не «патрулировали» данный сектор в поисках платформы, а быстро покидали его. В среднем время пребывания в данном секторе было значимо меньше, чем в группе без патологии.
Биохимический анализ. В табл. 4 приведены концентрации белка S100b в плазме крови. Проведение двухфакторного дисперсионного анализа с повторными измерениями [фактор 1 – «группа» (индукция патологии), фактор 2 – «время»] показал, что после гипогликемического состояния, вызванного введением инсулина, концентрация белка S100b в плазме крови статистически значимо увеличивалась и оставалась высокой, по сравнению с показателями в контрольной группе, на всем протяжении эксперимента (F1,10=43,3; р<0,001). Последующее межгрупповое сравнение подтвердило увеличение концентрации белка S100b в группе со сформированной патологией по сравнению с интактной группой во всех временных точках (р<0,01; критерий Бонферрони, факторы – «время» и «группа»;
см. табл. 4).
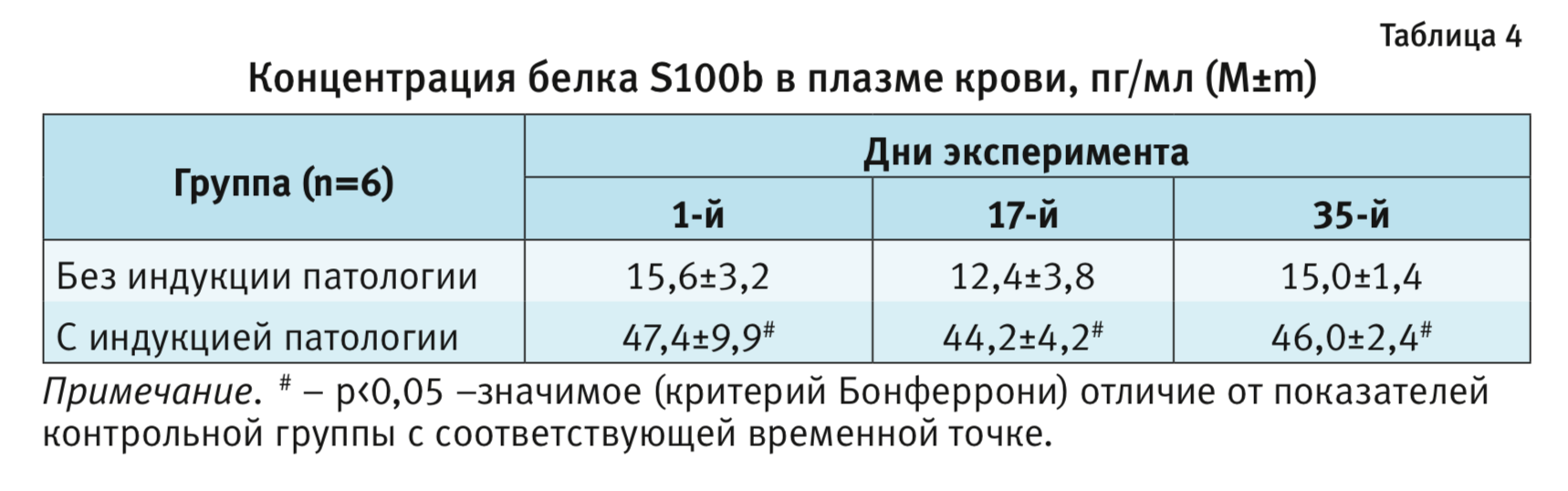
Таким образом, индукция патологии (гипогликемия) привела к увеличению концентрации белка S100b в плазме крови.
ИГХ анализ
Интенсивность ИГХ окрашивания S100b+ клеток в астроцитах и других глиальных клетках исследованной области гиппокампа (CA1) у животных группы с патологией значительно превышала данный показатель у контрольных крыс. На фоне развития патологии отмечалось увеличение количества иммунопозитивных клеток в поле зрения, а также внеклеточной экспрессии маркера (см. рисунок).
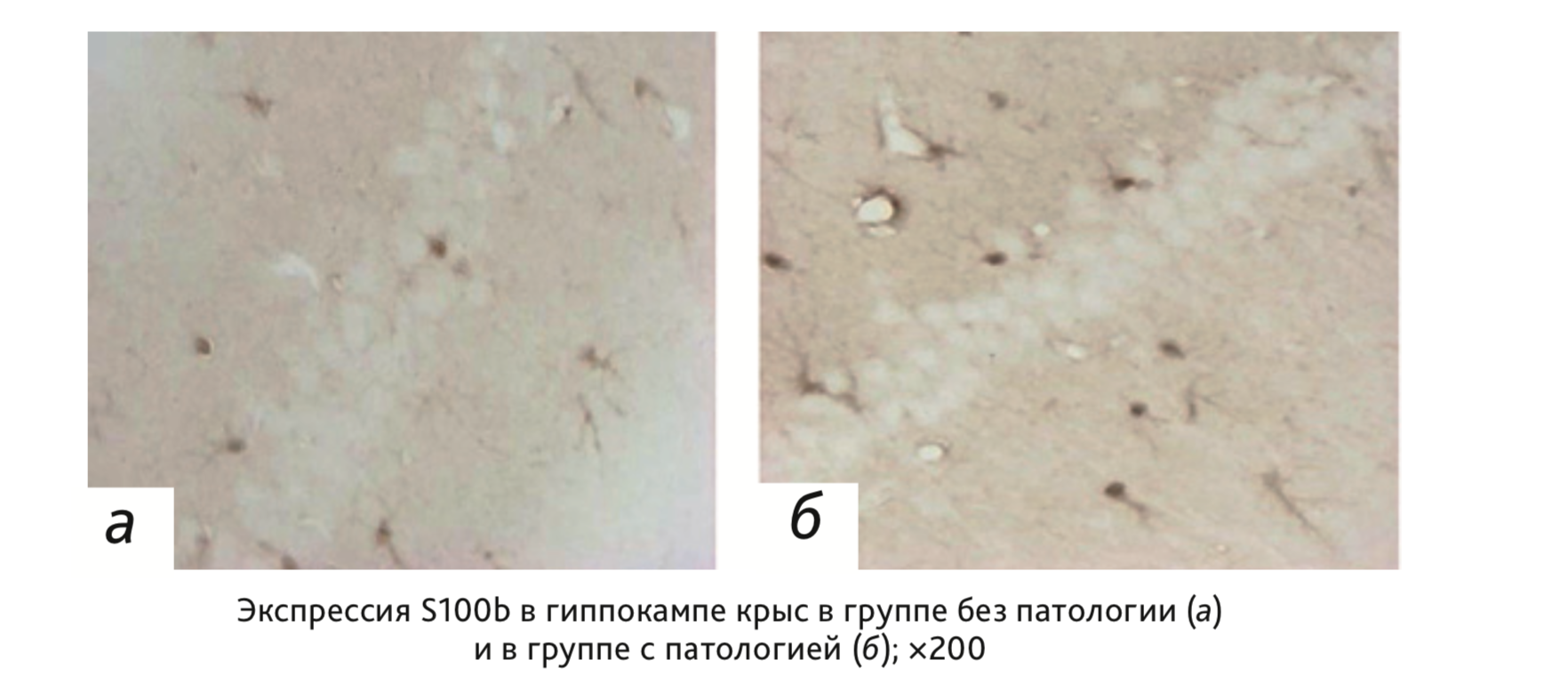
Интенсивность окрашивания иммунопозитивных клеток S100b через 35 дней после индукции патологии была значимо больше в гиппокампе крыс с патологией по сравнению с интактными животными (р<0,05; тест Манна–Уитни;
табл. 5).
При анализе данных экспрессии белка S100b по количеству иммунопозитивных клеток, метод парного сравнения показал увеличение количества положительно окрашенных клеток в гиппокампе через 35 дней после гипогликемии (р<0,05, t-тест, см. табл. 4).
Таким образом, продолжительная гипогликемия, вызванная введением инсулина у крыс, характеризовалась увеличением экспрессии белка S100b в гиппокампе к 35-му дню эксперимента.
Критическое снижение уровня глюкозы крови – часто встречающийся побочный эффект инсулиновой терапии больных сахарным диабетом (СД). Гипогликемией при проведении любой медикаментозной сахароснижающей терапии считается снижение уровня глюкозы ниже 3,3 ммоль/л. Снижение уровня глюкозы до уровня 1,7–2,6 ммоль/л приводит к уменьшению потребления глюкозы мозгом и энергетическому голоду нервных клеток, т.е. к нейрогликопении. Часто повторяющиеся гипогликемические состояния у человека рано или поздно приводят к необратимому поражению нейронов коры головного мозга, что клинически может проявляться церебрастенией, снижением интеллекта, эпилептиформными припадками [22]. Особенно опасна затянувшаяся (>30 мин) гипогликемическая кома [23]. В общей структуре смертности при СД гипогликемические комы составляют 3–4% [22].
В экспериментах используют модель развития когнитивных нарушений и морфологических изменений головного мозга в результате вызванной введением инсулина гипогликемии. Показано, что значительное снижение уровня глюкозы крови ведет к развитию отсроченных нарушений когнитивных функций, причиной которых могут быть вызванные гипогликемией повреждения нейронов головного мозга [1, 2].
В проведенном исследовании гипогликемическое состояние моделировали путем однократного подкожного введения инсулина в дозе 15 ЕД/кг крысам в течение 5 дней, с последующим выведением из патологического состояния введением 25% раствора глюкозы. Продолжительность периода гипогликемии составляла 60–70 мин. Средний уровень глюкозы в этот период был равным 2,1 ммоль/л.
Развитие когнитивного дефицита оценивалось по тесту «Водный лабиринт Морриса». Индукция гипогликемии привела к изменению паттерна поведения животных с индуцированной патологией, оно стало менее целенаправленным. Они чаще, чем контрольные животные посещали сектор, где в период обучения находилась платформа, но, в отличие от контрольных, не «патрулировали» данный сектор в поисках платформы, а быстро покидали его. Продолжительность пребывания в данном секторе была меньше по сравнению с показателями в контрольной группе. Таким образом, эпизоды умеренной гипогликемии привели к развитию когнитивных нарушений, возможно, обусловленных дефицитом внимания и/или нарушением процессов консолидации полученной на этапе обучения информации.
Оценка концентрации белка S100b в плазме крови показала, что непосредственно после формирования патологии и далее на протяжении всего наблюдения (до 35-го дня эксперимента) у животных, перенесших эпизоды гипогликемии, уровень белка был существенно повышен по сравнению с таковым в контрольной группе, что указывает на повреждение ткани головного мозга. Известно, что концентрация белка S100b при повреждениях нервной ткани увеличивается до микромолярной [15]. В данном исследовании повторяющиеся эпизоды умеренной гипогликемии у крыс также индуцировали увеличение концентрации S100b.
Кроме увеличения концентрации белка S100b в крови, наблюдалось увеличение его экспрессии в головном мозге. Было выявлено, что гипогликемия приводит к увеличению экспрессии белка S100b к 35-му дню эксперимента в зоне СА1 гиппокампа, что указывает на развитие индуцированных гипогликемическим состоянием нейротоксических процессов в данной структуре. Согласно полученным данным, повторные эпизоды гипогликемии приводят к снижению плотности синаптических контактов в результате повреждений дендритов нейронов гиппокампа, что предположительно может стать причиной когнитивных нарушений [12].
Известно, что эпизоды нейротоксического поражения головного мозга ведут к увеличению экспрессии апоптотического маркера каспаза-3, который является триггером активации микроглии [24, 25]. В течение 1-го дня после ишемического или гипоксического повреждения, микроглия претерпевает морфологические изменения, и достигает активированного амебоидного состояния, при этом происходит умеренное увеличения числа клеток. В подострой фазе (от 3 до 7 дней после эпизода) клетки микроглии в очагах поражения активизируются и становятся морфологически неотличимыми от инфильтрирующих макрофагов. Наличие смешанной популяция микроглии и макрофагов указывает на повреждение ГЭБ и массивную инфильтрацию циркулирующих иммунных клеток [25]. Через 1 нед после перенесенного ишемического или гипоксического эпизода в структурах головного мозга астроциты образуют своеобразный астроцитарный шрам, направленный к ишемической или гипоксической зоне поражения. Поскольку эпизоды гипогликемии приводят к активации микроглии как в гиппокампе, так и в коре головного мозга крыс, это подтверждает, что гипогликемические состояния инициируют и развитие нейровоспалительной реакции. Однако последний вклад в наблюдаемые при таких состояниях когнитивные нарушения пока не ясен [12].
Заключение
Таким образом, было показано, что эпизоды умеренной гипогликемии приводят к увеличению концентрации нейроспецифического белка S100b в плазме крови и коррелируют с увеличением экспрессии белка S100b в зоне СА1 гиппокампа. Следовательно, изменение концентрации данного белка является важным трансляционным и диагностическим маркером развития повреждений головного мозга, который можно оценивать в динамике. Данный подход может быть использован при изучении эффективности новых лекарственных препаратов.
Список источников
- Auer R. N. Hypoglycemic brain damage. Metab Brain Dis. 2004. Vol. 19; 3-4: 169–75.
- Suh S. W., Gum E. T., Hamby A. M., Chan P. H., Swanson R. A. Hypoglycemic neuronal death is triggered by glucose reperfusion and activation of neuronal NADPH oxidase. J. Clin. Invest. 2007. Vol. 117; 4: 910–8.
- Lindvall O., Auer R. N., Siesjo B. K. Mechanisms of hypoglycemic brain damage. Evidence against a significant role of the noradrenergic locus coeruleus system. Exp. Brain. Res. 1988. Vol. 73; 1: 219–23.
- Abdul-Rahman A., Siesjo B. K. Local cerebral glucose consumption during insulin-induced hypoglycemia, and in the recovery period following glucose administration. Acta Physiol. Scand. 1980. Vol. 110; 2: 149–59.
- Langan S. J., Deary I. J., Hepburn D. A., Frier B. M. Cumulative cognitive impairment following recurrent severe hypoglycaemia in adult patients with insulin-treated diabetes mellitus. Diabetologia. 1991. Vol. 34; 5: 337–44.
- Suh S. W., Aoyama K., Chen Y., Garnier P., Matsumori Y., Gum E., Liu J., Swanson R. A. Hypoglycemic neuronal death and cognitive impairment are prevented by poly(ADP-ribose) polymerase inhibitors administered after hypoglycemia. J. Neurosci. 2003. Vol. 23; 33: 10681–90.
- Suh S. W., Fan Y., Hong S. M., Liu Z., Matsumori Y., Weinstein P. R., Swanson R. A., Liu J. Hypoglycemia induces transient neurogenesis and subsequent progenitor cell loss in the rat hippocampus. Diabetes. 2005. Vol. 54; 2: 500–9.
- Wieloch T., Engelsen B., Westerberg E., Auer R. Lesions of the glutamatergic cortico-striatal projections in the rat ameliorate hypoglycemic brain damage in the striatum. Neurosci Lett. 1985. Vol. 58; 1: 25–30.
- Bree A. J., Puente E. C., Daphna-Iken D., Fisher S. J. Diabetes increases brain damage caused by severe hypoglycemia. Am. J. Physiol. Endocrinol. Metab. 2009. Vol. 297; 1: E194–201.
- Nellgard B., Wieloch T. Cerebral protection by AMPA- and NMDA-receptor antagonists administered after severe insulin-induced hypoglycemia. Exp. Brain. Res. 1992. Vol. 92; 2: 259–66.
- Auer R. N., Olsson Y., Siesjo B. K. Hypoglycemic brain injury in the rat. Correlation of density of brain damage with the EEG isoelectric time: a quantitative study. Diabetes. 1984. Vol. 33; 11: 1090–8.
- Won S. J., Yoo B. H., Kauppinen T. M., Choi B. Y., Kim J. H., Jang B. G., Lee M. W., Sohn M., Liu J., Swanson R. A., Suh S. W. Recurrent/moderate hypoglycemia induces hippocampal dendritic injury, microglial activation, and cognitive impairment in diabetic rats. J. Neuroinflammation. 2012. Vol. 9: 182.
- Languren G., Montiel T., Julio-Amilpas A., Massieu L. Neuronal damage and cognitive impairment associated with hypoglycemia: An integrated view. Neurochemistry International. 2013. Vol. 63; 4: 331–43.
- Morris R. Developments of a water-maze procedure for studying spatial learning in the rat. J. Neurosci. Methods. 1984. Vol. 11; 1: 47–60.
- Sorci G., Bianchi R., Riuzzi F., Tubaro C., Arcuri C., Giambanco I., Donato R. S100B Protein, A Damage-Associated Molecular Pattern Protein in the Brain and Heart, and Beyond. Cardiovasc. Psychiatry. Neurol. 2010. Vol. 2010.
- Languren G., Montiel T., Ramírez-Lugo L., Balderas I., Sánchez-Chávez G., Sotres-Bayón F., Bermúdez-Rattoni F., Massieu L. Recurrent moderate hypoglycemia exacerbates oxidative damage and neuronal death leading to cognitive dysfunction after the hypoglycemic coma. Journal of Cerebral Blood Flow & Metabolism. 2017: 0271678X17733640.
- Коптяева К. Е., Мужикян А. А., Гущин Я. А., Беляева Е. В., Макарова М. Н., Макаров В. Г. Некоторые особенности фиксации органов и тканей лабораторных животных для повышения качества гистологического анализа. Лабораторные животные для научных исследований. 2018. Vol. 2: 60–70.
- Коптяева К. Е., Мужикян А. А., Гущин Я. А., Беляева Е. В., Макарова М. Н., Макаров В. Г. Методика вскрытия и извлечения органов лабораторных животных (крысы). Лабораторные животные для научных исследований. 2018. Vol. 2^ 71–93.
- Swanson L.W. Back cover of Brain Maps, third edition Электронный ресурс: http://larrywswanson.com/?page_id=164
- Мужикян А. А., Макарова М. Н., Гущин Я. А. Особенности гистологической обработки органов и тканей лабораторных животных/ Международный вестник ветеринарии. 2014. Vol. 2: 103–9.
- Buesa R. J., Peshkov M. V. Histology without xylene. Ann. Diagn. Pathol. 2009. Vol. 13; 4: 246–56.
- Сахарный диабет (учебное пособие). Родионова Т. И. Саратов, 2007.
- Алгоритмы специализированной помощи больным сахарным диабетом. Дедов И. И. М., 2013.
- Burguillos M. A., Deierborg T., Kavanagh E., Persson A., Hajji N., Garcia-Quintanilla A., Cano J., Brundin P., Englund E., Venero J. L., Joseph B. Caspase signalling controls microglia activation and neurotoxicity. Nature. 2011. Vol. 472; 7343: 319–24.
- Yoon J. S., Jo D., Lee H. S., Yoo S. W., Lee T. Y., Hwang W. S., Choi J. M., Kim E., Kim S. S., Suh-Kim H. Spatiotemporal Protein Atlas of Cell Death-Related Molecules in the Rat MCAO Stroke Model. Exp. Neurobiol. 2018. Vol. 27; 4: 287–298.
Поиск



