
Muc2-дефицитные мыши как модель хронического воспаления
Работа выполнена без спонсорской поддержки.
Милутинович К.С., Попов В.С. Muc2-дефицитные мыши как модель хронического воспаления. Лабораторные животные для научных исследований. 2023; 3. https://doi.org/10.57034/2618723X-2023-03-11
Резюме
Воспалительные заболевания кишечника (ВЗК) — это группа заболеваний, развитию которых способствует широкий ряд факторов, а существующее лечение направлено на купирование симптомов. Характерной чертой ВЗК является воспаление слизистой оболочки кишечника и последующее возникновение воспалительных процессов, что приводит к нарушению пищеварения. Нарушение слизистого барьера приводит к развитию патологий, в результате активируется локально иммунная система, и развивается воспаление. В ответ на воспаление, проникающее в ткань кишечника, макрофаги поляризуются по провоспалительному типу, секретируя провоспалительные цитокины. В результате развивается хроническое воспаление, со временем способствующее развитию осложнений, в том числе опухолей. Схожий патогенез развития ВЗК наблюдается у животной модели ВЗК — мышей с нокаутом гена муцина 2. У мышей Muc2-/- истощен слизистый слой толстого кишечника, в норме формируемый белком муцина 2. Отсутствие нормального слизистого барьера приводит к развитию хронического воспаления, по симптоматике и проявлениям схожего с таковым у пациентов с ВЗК. Немаловажной характеристикой данной модели ВЗК является возможность изучать широкий спектр симптомов на одной модели без влияния искусственных индукторов. Такие особенности мышей с нокаутом гена муцина 2 делают эту линию мышей не только эффективной моделью ВЗК, но и моделью хронических воспалительных процессов в целом.
Введение
Воспалительные заболевания кишечника (ВЗК) — широко распространенная группа хронических заболеваний [1], встречаемость которых достигает 369 случаев на 100 000 населения [2]. На сегодняшний день непосредственные и отдаленные причины возникновения ВЗК активно исследуются, существующие подходы к терапии нельзя признать удовлетворительными. Так, применение антибиотиков, аминосалицилатов, кортикостероидов и иммуносупрессантов нельзя считать достаточно эффективным методом лечения, поскольку данные препараты снижают лишь симптомы ВЗК. Именно поэтому так важен поиск новых методов лечения, продолжающийся и по сей день [3, 4].
Исследование новых терапевтических подходов, действующих на фундаментальные механизмы развития ВЗК, основным из которых является воспаление, предполагает расширение арсенала исследовательских моделей, воспроизводящих ключевые симптомы. При выборе животной модели для исследования ВЗК трансгенные модели имеют преимущество, поскольку воспроизводят картину патологии, развивающейся в течение всей жизни, с учетом возможных компенсаторных механизмов.
Цель работы — изучить и объединить имеющиеся данные об уникальной модели ВЗК, мышах с нокаутом гена муцина (Muc2-/-), созданной в Нидерландах, а также обосновать эффективность использования Muc2-/- мышей в качестве модели хронического воспаления.
Поиск литературы осуществляли в базах PubMed и Goggle Scholar.
Патогенез воспалительных заболеваний кишечника
ВЗК — это хронические иммуновоспалительные заболевания [1]. Самые распространенные случаи ВЗК — это болезнь Крона и язвенный колит. В большинстве своем ВЗК развивается у человека в возрасте до 30 лет. ВЗК проявляются возникновением хронического воспаления на протяжении всего кишечного тракта [5]. Для ВЗК также характерны периоды длительной ремиссии с рецидивами и многочисленные внекишечные осложнения [1]. Однако точная этиология ВЗК до сих пор обсуждается [5].
Болезнь Крона отличается от язвенного колита локализацией воспаления: язвенный колит в основном поражает отделы толстого кишечника, в то время как при болезни Крона поражается в основном тонкий кишечник. У пациентов с язвенным колитом часто наблюдается ректальное кровотечение, чего почти не наблюдается у пациентов с болезнью Крона, но у последних часто возникают осложнения в виде свищей [5, 6].
Ключевым симптомом развития язвенного колита является диффузное хроническое воспаление слизистой оболочки, распространяющееся проксимально от прямой кишки. Причиной воспаления служит нарушение слизистого слоя, вызванное истощением муцина 2 — основного секреторного муцина бокаловидных клеток толстого кишечника [7, 8].
Показано, что нарушение слизистого слоя приводит к контакту кишечной флоры с эпителиальными тканями толстой кишки [9], в результате чего активируется локальная иммунная система [10, 11]. Проникновению бактерий в слой эпителия кишечника способствует также нарушение регуляции плотных контактов эпителия, что становится причиной распространения воспалительного процесса не только на слизистую оболочку, но и на подлежащие слои вплоть до мышечного [12–14]. В результате активации врожденного иммунитета [15] происходит привлечение Т-хелперов, нейтрофилов и макрофагов в места воспаления [16]. При этом макрофаги не только секретируют широкий спектр цитокинов, запуская воспалительный процесс, но также и принимают участие в регуляции восстановления поврежденной ткани [14].
При запуске воспалительных процессов макрофаги моноцитарного происхождения первыми из иммунных клеток проникают в ткани кишечника [17]. При этом происходит поляризация макрофагов по провоспалительному типу [18], и они секретируют провоспалительные цитокины [14]. Так, макрофаги продуцируют CD14, TREM-1 и человеческий миелоидный IgA Fc-рецептор CD89, активированный NF-κB [18], TNF, IL-6, IL-8, IL-23, IL-1β и IFNγ и хемокины (CCL2) [19].
При исследовании роли макрофагов кишечника в патогенезе ВЗК было показано, что у мышей с дефицитом IL-10 макрофаги в ответ на контакт с комменсальной флорой способствуют развитию опосредованного Т-хелперами хронического колита за счет продукции IL-12 и IL-23 [18].
Клеточный профиль иммунного ответа у пациентов с ВЗК имеет специфические особенности. У больных ВЗК наблюдается значительное снижение количества CD103+-дендритных клеток [9], отвечающих за регуляцию иммунного ответа [20], в то время как количество CD14+-макрофагов, участвующих в распознавании бактерий [17], наоборот, возрастало [9]. Такие изменения в составе иммунных клеток отражают аутоиммунную реакцию на собственную микрофлору. Воспаление, возникающее в кишечнике, сопровождается увеличением синтеза антибактериальных молекул и изменением регуляции активности иммунных клеток [21]. Наряду с этим возрастает продукция провоспалительных цитокинов, в том числе TNF-α [22], IL-1 β, IL-6, IL-12, IL-23 и хемокинов [23].
Значение интерлейкинов для патогенеза ВЗК может быть проиллюстрировано на примере IL-23. Известно, что ряд мутаций в гене рецептора IL-23 (IL23R) способствует развитию ВЗК. В то же время увеличение продукции IL-23 способствует пролиферации Т-клеток кишечника, накоплению Т-хелперов-17 [24, 25], участвующих в аутоиммунных реакциях [26], а также снижает дифференцировку Foxp3+ Т-клеток, способствующих снижению иммунного ответа, и как следствие — секрецию IL-10 [24, 25]. Снижение экспрессии гена IL-10 [27] или его рецепторов в свою очередь ассоциировано с развитием колита [25]. Многоцентровое исследование антитела-антагониста к рецептору IL-6 показало клиническое улучшение у пациентов с ВЗК, подтвердив значение этого сигнального пути как терапевтической мишени [28].
Кроме того, известно, что ряд полиморфизмов в генах секреторных молекул дендритных клеток увеличивает вероятность развития ВЗК. К таким генам относится ген цитокина семейства TNF Tl1α, повышенная экспрессия которого наблюдалась у пациентов [25].
Наличие у пациентов ВЗК повышает риск онкологических осложнений, так как хроническое воспаление способствует развитию опухоли путем активации пролиферации и противоапоптозных механизмов предраковых клеток, а также прогрессированию опухоли и метастазов при ее возникновении. Хемокины не только регулируют миграцию инвазивных клеток к кровеносным сосудам, но некоторые, такие как CXCR4, CCR4, CCR7, CCR9 и CCR10, также контролируют миграцию метастатических клеток в отдаленные органы. Провоспалительные цитокины, такие как IL-6 и IL-11, контролируют экспрессию фактора семейства Tff3, что также может способствовать метастазированию [28].
Таким образом, именно макрофагальное звено иммунного ответа является ключевым регулятором развития патогенеза ВЗК и остроты воспаления. Определяющими процессами при этом выступают поляризация макрофагов по провоспалительному типу и паракринная регуляция с помощью синтеза провоспалительных цитокинов. Поскольку ВЗК характеризуются активной пролиферацией кишечного эпителия в онкогенных условиях, образование опухолей является одним из тяжелых осложнений.
Причины возникновения ВЗК и подходы к терапии
Причины развития ВЗК многочисленны. Известно, что внешняя среда может способствовать развитию ВЗК. К негативным факторам внешней среды относят диету, географическое положение, социальный стресс, а также наркотики и курение [29, 30]. Не менее важен вклад в патогенез ВЗК дисфункции врожденных и адаптивных иммунных путей, в том числе в результате генетических нарушений [29]. Установлено, что наиболее значимым фактором для развития ВЗК является генетическая предрасположенность и наследственность. В настоящее время показано, что генетическая предрасположенность может влиять на барьерную функцию кишечника и работу иммунной системы [7]. Выявлено более 200 мутаций, коррелирующих с возникновением ВЗК [31].
Формированию ВЗК может способствовать нарушение бактериального состава кишечной флоры у пациентов [32], что приводит к изменению соотношения бактериальной микрофлоры кишечника. Это является крайне важным аспектом для развития воспаления [25]. У пациентов с болезнью Крона наблюдалось снижение количества ряда симбиотических бактерий, таких как Firmicutes и Bacteroidetes [25, 33, 34]. В случае ВЗК, вызванных дефицитом слизистого слоя, отсутствие муцина 2, а вместе с ним и терминальной молекулы муцина 2 фукозы, может способствовать устойчивости патогенов в кишечнике человека [35].
Главной целью, преследуемой при лечении ВЗК, является улучшение качества жизни пациентов посредством переключения заболевания с активной формы на ремиссию и ее поддержание [36]. В настоящее время применяется ряд препаратов для купирования симптомов ВЗК. К ним относятся кортикостероиды, 5-аминосалициловая кислота, иммуномодуляторы и антибиотики. В некоторых случаях при лечении ВЗК используют сорбирующие средства и антибиотики, что помогает нормализации пищеварительных процессов [37]. Также в настоящее время идет поиск наиболее эффективных молекул, в том числе факторов роста, способствующих регенерации слизистого слоя в кишечнике [38]. Однако все эти подходы не излечивают пациентов, а лишь купируют симптомы заболеваний [5], обеспечивая в лучшем случае долговременную ремиссию. К тому же в некоторых случаях указанная терапия выступает как стимул для развития ряда тяжелых осложнений [39]. Многочисленные данные свидетельствуют о резистентности части пациентов к терапии или снижении эффективности лечения со временем [40], поэтому так важен поиск новых методов лечения.
Роль муцина 2 в патогенезе ВЗК
Выше мы упоминали о нарушении целостности слизистого слоя кишечника как ключевом событии развития хронического воспаления. Основными компонентами слизистого слоя толстого кишечника являются муцины, в первую очередь муцин 2 [41]. Белок муцина 2 обладает тандемными, нерегулярными повторяющимися последовательностями, а также богат треониновыми и сериновыми участками, необходимыми для о-гликозилирования [42]. Сайты о-гликозилирования могут служить местом адгезии для бактерий, а также служат источником энергии для симбиотических бактерий [43].
Муцин 2 синтезируется бокаловидными клетками, в которых он подвергается процессингу и хранится в секреторных гранулах до момента секреции, после чего распределяется по кишечному тракту [14, 41]. Однако это распределение неоднородно: в толстом кишечнике имеются два разных по плотности слоя слизи [44], в то время как в тонком кишечнике есть только один рыхлый слой [14]. Внутренний слой толстого кишечника, наиболее твердый и плотный, обладает высокой концентрацией муцина 2. Внутренний слой имеет маленькие поры, что блокирует попадание бактерий в этот слой [43]. Внешний слой рыхлый, является средой обитания комменсальных бактерий [44]
Таким образом, слизистый слой кишечника имеет два слоя: защитный внутренний, в норме непроницаемый для симбиотических бактерий и патогенов, и внешний, предоставляющий им среду обитания [21, 45]. Внутренний слой слизи обеспечивает первую линию защиты при развитии ВЗК 9 (рис. 1) [46].
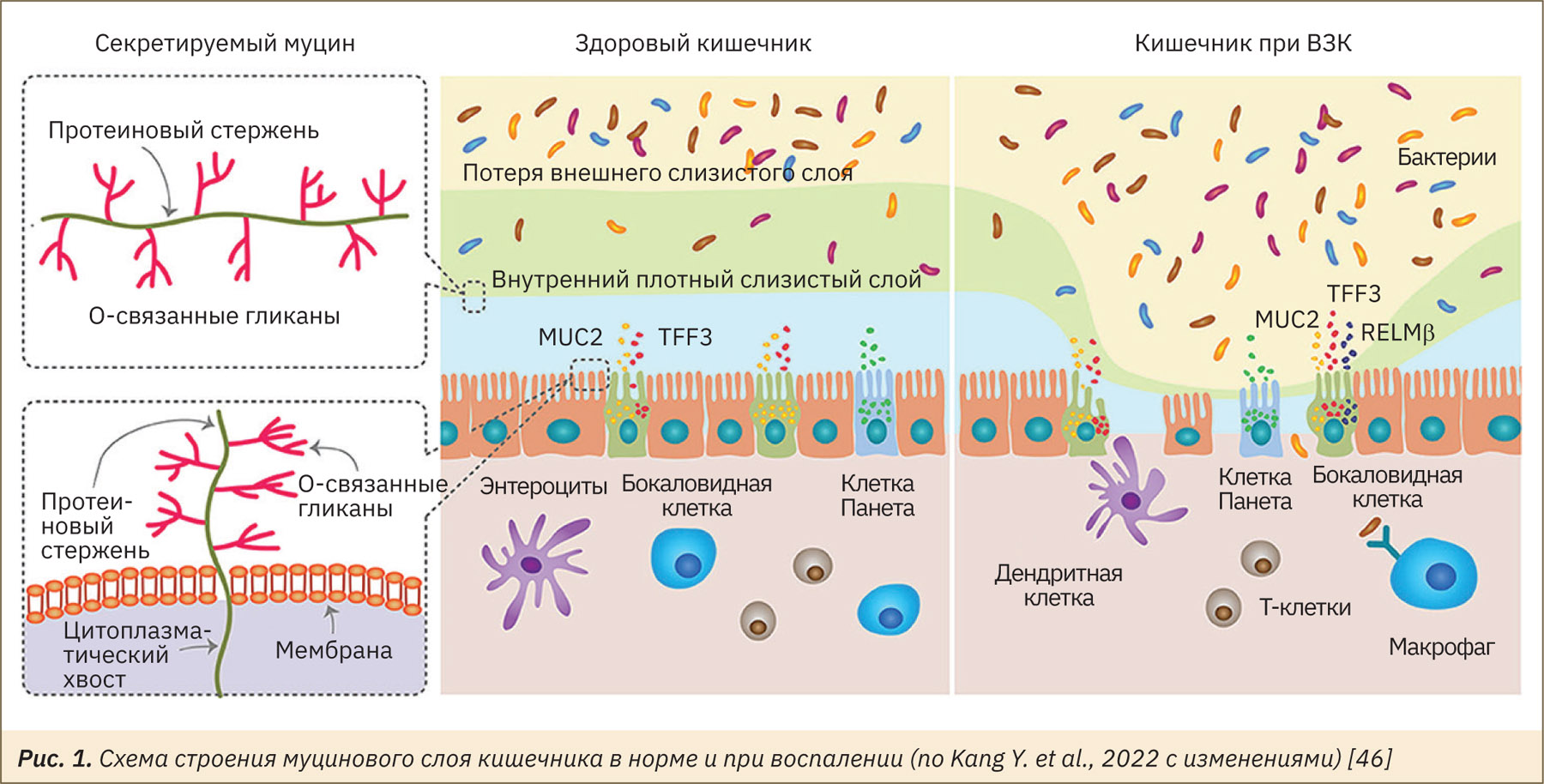
Терминальная молекула муцина 2 — моносахарид фукозы, участвующий во взаимодействиях с белками [47]. Фукоза является источником пищи для бактерий кишечника, а также может подавлять вирулентность патогенов в кишечнике хозяина [35]. В последнее время фукозе придается важное значение как потенциальному агенту, снижающему агрессивное действие микробного сообщества кишечника при ВЗК [48, 49]. Была показана связь гликозилирования муцина 2 и ВЗК: у пациентов терминальные участки молекулы представлены более короткими гликанами [50].
Таким образом, муцин 2 — основной структурный компонент кишечной слизи, необходимый для выполнения ею защитной функции [51]. Участие муцина 2 в формировании эффективного физического барьера и препятствие адгезии патогенных микроорганизмов крайне необходимы для защиты кишечного эпителия [52].
Потеря муцина 2 приводит к истощению слизистого слоя, что в свою очередь вызывает развитие воспалительных процессов, характерных для ВЗК [53].
Нокаутные по муцину 2 мыши как модель ВЗК
Многообразие причин, приводящих к ВЗК, и сложность терапии этих заболеваний вызывают потребность в различных моделях для разработки лекарств. Наряду с моделями индуцированного ВЗК, к которым относятся модель, индуцируемая декстрансульфатом натрия (DSS) [54], модели, индуцируемые 2,4,6-тринитробензолсульфокислотой (TNBS) и динитробензолсульфокислотой (DNBS) [55], а также модели с переносом популяции Т-клеток иммуносупрессированным мышам [54], используются трансгенные мыши с нокаутом IL-10-/- [56] или с нокаутом IL-23-/- [20] и др. [55].
Одной из используемых моделей воспалительных заболеваний кишечника является модель мышей с нокаутом по гену муцина 2 (Muc2-/-), у которых спонтанно развивается язвенный колит [9].
Наиболее важной характеристикой данной модели ВЗК является возможность изучать широкий спектр симптомов ВЗК на одной модели. Тем самым складывается наиболее точное представление о патогенезе ВЗК. Более того, симптомы ВЗК у мышей с нокаутом муцина 2 развиваются спонтанно, само заболевание протекает с длительными периодами ремиссии, что также свойственно и для пациентов. Отсутствие необходимости искусственно формировать патологическое состояние позволяет изучать непосредственно причинно-следственные связи, лежащие в основе самого заболевания, а не комбинации симптомов и побочных эффектов индукции ВЗК.
Изучаемые мыши с нокаутом гена муцина 2 впервые были созданы в Нидерландах А. Velcich [57]. Нокаут гена был получен на базе C57BL/6J 129/SvOla линий мышей путем замены геномного фрагмента, охватывающего экзоны 2–4-го гена муцина 2, на фрагмент, содержащий ген, определяющий устойчивость к неомицину, под контролем промотора фосфоглицераткиназы-1 (PGK-Neo). Изначально мыши были созданы для изучения колоректального рака. Начиная с 6-го месяца жизни у них формируются аденокарциномы по всей длине кишечника, к 1 году опухоли образуются у 100% нокаутных мышей [51].
У мышей с нокаутом гена муцина 2 развивается недостаточность слизистого слоя, в результате бактерии входят в контакт непосредственно с эпителиальными клетками кишечника [9, 43]. Показано, что у мышей с нокаутом муцина 2 увеличивается проницаемость кишечника [58], которая приводит к попаданию в кровь бактерий и вызывает интоксикацию [59]. Тесный контакт бактерий с эпителиальной стенкой кишечника способствует развитию хронических воспалительных реакций и гиперплазии эпителия, что в конечном итоге приводит к формированию опухолей [43]. Таким образом, эта линия мышей воспроизводит патогенетическую цепочку, характерную для пациентов с ВЗК [8, 46].
Муцин 2 является основой слизистой оболочки кишечного тракта как у человека, так и у мышей [60]. Отсутствие столь важной молекулы влечет за собой нарушения морфологии тканей преимущественно в дистальных отделах желудочно-кишечного тракта, где содержание муцинов в слизистом слое наибольшее. Это приводит и к функциональным нарушениям. Так, с рождения мышата набирают меньшую массу тела из-за недоедания [61], в период с 8-й по 20-ю неделю у животных начинает спонтанно возникать колит [9], а с 6-го месяца у мышей формируются опухоли [51].
Характерными симптомами ВЗК у мышей с дефицитом муцина 2 являются снижение массы тела, наличие кишечных кровотечений с 8-й недели жизни, а также ректальный пролапс. Начиная с 9-й недели жизни у некоторых мышей наблюдается более жидкий стул [53]. Чтобы точнее оценить симптомы, используют систему Disease activity index (DAI), где с помощью балльной схемы оценивается тяжесть клинических проявлений у мышей [35]. По шкале от 1 до 4 оцениваются наличие или отсутствие, а также выраженность следующих симптомов: потеря массы тела, нарушение стула, ректальное кровотечение [61, 62].
Интересно, что у мышат вплоть до 28-го дня жизни, момента перехода с материнского вскармливания на полноценный рацион, не наблюдается значимых изменений в морфологии кишечника, за исключением отсутствия типичной колоколообразной формы бокаловидных клеток [63]. После перехода на самостоятельное питание начинают выявляться патологические изменения в кишечнике: с 16-недельного возраста у мышей Muc2-/- детектировались увеличение длины крипт, а также потеря архитектуры крипт и общей структуры собственной пластинки в проксимальном отделе толстой кишки. В дистальном отделе толстого кишечника уже с 5-й недели наблюдаются гиперплазия крипт, уплощение эпителиальных клеток и инфильтрация иммунных клеток. Начиная с 12–16-й недели жизни в дистальном отделе перестают детектироваться бокаловидные клетки. К 16-й неделе заметно уплощается и искажается структура эпителия, сильно увеличивается длина крипт, а также наблюдается активная инфильтрация воспалительных клеток и эпителиальная эрозия [53, 64].
При гистологическом анализе у животных с нокаутом гена муцина 2 почти не детектируются бокаловидные клетки в срезах двенадцатиперстной кишки, а также на протяжении всех отделов толстой кишки мышей. Однако экспрессия трефоидного фактора (Itf) [63], являющегося маркером дифференцированных бокаловидных клеток [51], увеличивается, что может быть следствием удлинения крипт [63].
При исследовании миграции эпителиальных клеток кишечника выяснили, что эпителиальные клетки кишечника мышей Muc2-/- мигрировали намного быстрее в просвет кишечника по сравнению с клетками мышей Muc2+/+. Такое наблюдение может быть следствием бактериальной стимуляции эпителия кишечника и действием на клетки провоспалительных цитокинов [51, 53].
Исследования цитокинов выявили значительное увеличение (в 5 раз) количества мРНК воспалительного цитокина TNF-α в толстой кишке мышей Muc2-/- по сравнению с контрольными. Кроме того, мыши Muc2-/- продемонстрировали тенденцию к усилению (в 4 раза) экспрессии гена IL-1β в толстой кишке [53].
При исследовании влияния экзогенной фукозы на состояние мышей Muc2-/- обнаружено, что уровень АТФ у перитонеальных макрофагов нокаутных мышей был значительно снижен по сравнению с животными контрольной группы мышей C57Bl6. Это может быть следствием возникновения окислительного стресса у мышей Muc2-/-, что также характерно и для пациентов с ВЗК [64].
Отсутствие нормальной слизистой оболочки у нокаутных по муцину 2 мышей приводит к развитию воспалительного процесса, симптоматика которого схожа с таковой у пациентов с язвенным колитом [9]. Основными цитокинами, увеличение секреции которых наблюдается при ВЗК у пациентов, являются TNF-α, IL-1β и IL-6 [53]. Кроме того, изменяется экспрессия IL-12 и INF-γ [65].
Схожим образом у мышей Muc2-/- обнаружено повышение экспрессии TNF-β и IL-1β [52, 66]. У мышей также была увеличена экспрессия интерлейкина-1-альфа (IL-1α) в сравнении с мышами C57BL/6 и экспрессия транскрипционного фактора Т-регуляторных клеток (Foxp3), в то время как уровень экспрессии транскрипционных факторов Т-хелперов-1 (Th1) и Т-хелперов-17 (Th17), Tbx21 и Rorc соответственно не отличался от контрольной группы C57BL/6. Не обнаружено различий и в экспрессии цитокинов (IL-22, IL-17, IL-12p40, IL-12p70), участвующих в Т-хелперном опосредованном ответе [49].
F. Obermeier и соавт. [67] в своей работе на разных моделях ВЗК продемонстрировали роль толл-подобных рецепторов (TLR) при хроническом воспалении, когда их экспрессия значительно увеличена. У мышей Muc2-/- на фоне перехода на самостоятельное питание начинает формироваться воспаление, в результате перестают экспрессироваться TLR, а также перестраивается экспрессия цитокинов. TLR4- и TLR9-рецепторы предположительно вносят вклад в ограничение развития колита в период грудного вскармливания. Однако к 28-му дню жизни у мышат начинают формироваться первые признаки воспаления. Происходит увеличение экспрессии генов Cd45, Cd3ε и Foxp3, маркеров Т-клеток в сочетании с повышением экспрессии генов цитокинов IL-12, p35, IL-10, TGF-β1 и TNF-α в дистальном отделе толстого кишечника. У мышат на 28-й день жизни также изменяется морфология кишечника, появляются первые признаки уплощения эпителия и эрозия, в то время как в проксимальном отделе таких изменений не наблюдается [63].
Заключение
Таким образом, мыши, нокаутные по гену муцина 2, хорошо воспроизводят и симптоматику, и патогенетическую картину, наблюдаемую при развитии воспалительного заболевания кишечника у человека. В отличие от индуцируемых моделей у нокаутных мышей наблюдается развитие патологии, повторяющей характеристики патологического процесса у пациентов, — спонтанное возникновение, плавное развитие, длительные ремиссии. Важным является и гарантированное развитие опухолей как следствие воспалительного процесса, то есть ясный финал с хорошо исследованным таймингом. Перечисленные особенности делают эту линию мышей очень удобной моделью для исследования новых терапевтических подходов к лечению воспалительных заболеваний кишечника и хронических воспалительных процессов в целом.
Сведения о конфликте интересов
Авторы заявляют об отсутствии конфликта интересов.
Вклад авторов
К.С. Милутинович — анализ данных научной литературы, написание и редактирование
текста рукописи.
В.С. Попов — редактирование текста и одобрение окончательного варианта
рукописи для публикации.
Список источников
-
Knyazev O.V. et al. Epidemiology of inflammatory bowel disease. State of the problem (review) // Dokazatel’naya Gastroenterol. Media Sphere Publishing Group. 2020. Vol. 9. N. 2. P. 66.
-
Alatab S. et al. The global, regional, and national burden of inflammatory bowel disease in 195 countries and territories, 1990–2017: a systematic analysis for the Global Burden of Disease Study 2017 // Lancet Gastroenterol. Hepatol. 2020. Vol. 5. P. 17–30.
-
Liu F. et al. Mechanistic insights into the attenuation of intestinal inflammation and modulation of the gut microbiome by krill oil using in vitro and in vivo models // Microbiome. BioMed Central Ltd. 2020. Vol. 8. N. 1. P. 1–21.
-
Fantini M.C., Guadagni I. From inflammation to colitis-associated colorectal cancer in inflammatory bowel disease: Pathogenesis and impact of current therapies // Dig. Liver Dis. W.B. Saunders. 2021. Vol. 53. N. 5. P. 558–565.
-
Walfish A.E., Companioni R.A.C. Overview of Inflammatory Bowel Disease // Gastrointestinal Disorders — MSD Manual Professional Edition. 2022.
-
Ивашкин В.Т. и др. Проект клинических рекомендаций по диагностике и лечению язвенного колита // Колопроктология. 2019. Т. 18. № 4. С. 7–36. [Ivashkin V.T. et al. Proekt klinicheskix rekomendacij po diagnostike i lecheniyu yazvennogo kolita // Koloproktologiya. 2019. Vol. 18. N. 4. P. 7–36. (In Russ.)].
-
Xavier R.J., Podolsky D.K. Unravelling the pathogenesis of inflammatory bowel disease // Nature. Nature Publishing Group, 2007. Vol. 448. N. 7152. P. 427–434.
-
Johansson M.E.V. et al. Bacteria penetrate the normally impenetrable inner colon mucus layer in both murine colitis models and patients with ulcerative colitis // Gut. BMJ Publishing Group. 2014. Vol. 63. N. 2. P. 281–291.
-
Wenzel U.A. et al. Spontaneous colitis in Muc2-deficient mice reflects clinical and cellular features of active ulcerative colitis // PLoS One. 2014. Vol. 9. N. 6.
-
Barbara G. et al. The Intestinal Microenvironment and Functional Gastrointestinal Disorders // Gastroenterology. 2016. Vol. 150. N. 6. P. 1305–1318. e8.
-
Vialov S.S. Mucosal permeability disturbances as a pathogenesis factor of gastrointestinal tract functional disorders: rationale and correction possibilities // Cons. Medicum. MediaMedica. 2018. Vol. 20. N. 12. P. 99–104.
-
Plichta D.R. et al. Therapeutic Opportunities in Inflammatory Bowel Disease: Mechanistic Dissection of Host-Microbiome Relationships // Cell. 2019. Vol. 178. N. 5. P. 1041–1056.
-
Simanenkov V.I. et al. Syndrome of increased epithelial permeability in clinical practice. Multidisciplinary national Consensus // Cardiovasc. Ther. Prev. Russian Fed. Vserossiiskoe Obshchestvo Kardiologov. 2021. Vol. 20. N. 1. P. 121–278.
-
Zhang Y. et al. ECM1 is an essential factor for the determination of M1 macrophage polarization in IBD in response to LPS stimulation // Proc. Natl. Acad. Sci. U.S.A. National Academy of Sciences. 2020. Vol. 117. N. 6. P. 3083–3092.
-
Mills C.D. M1 and M2 Macrophages: Oracles of Health and Disease // Crit. Rev. Immunol. 2012. Vol. 32. N. 6. P. 463–488.
-
Wirtz S. et al. Chemically induced mouse models of acute and chronic intestinal inflammation // Nat. Protoc. 2017. Vol. 12. N. 7. P. 1295–1309.
-
Kühl A.A. et al. Diversity of Intestinal Macrophages in Inflammatory Bowel Diseases // Front. Immunol. 2015. Vol. 6. N. DEC.
-
Kamada N. et al. Unique CD14 intestinal macrophages contribute to the pathogenesis of Crohn disease via IL-23/IFN-gamma axis // J. Clin. Invest. 2008. Vol. 118. N. 6. P. 2269–2280.
-
Monteleone G. et al. Blocking Smad7 restores TGF-beta1 signaling in chronic inflammatory bowel disease // J. Clin. Invest. 2001. Vol. 108. N. 4. P. 601–609.
-
Sennikov S.V. et al. Molecular and cellular mechanisms mediated by dendritic cells involved in the induction of tolerance // Med. Immunol. 2017. Vol. 19. N. 4. P. 359–374.
-
Johansson M.E.V., Hansson G.C. Immunological aspects of intestinal mucus and mucins // Nat. Rev. Immunol. NIH Public Access. 2016. Vol. 16. N. 10. P. 639.
-
Schierova D. et al. Fecal microbiome changes and specific anti-bacterial response in patients with ibd during anti-tnf therapy // Cells. MDPI. 2021. Vol. 10. N. 11.
-
Abraham C., Cho J.H. Inflammatory Bowel Disease // N. Engl. J. Med. NIH Public Access. 2009. Vol. 361. N. 21. P. 2066.
-
Ahern P.P. et al. Interleukin-23 Drives Intestinal Inflammation through Direct Activity on T Cells // Immunity. Cell Press, 2010. Vol. 33. N. 2. P. 279–288.
-
Bates J., Diehl L. Dendritic cells in IBD pathogenesis: an area of therapeutic opportunity? // J. Pathol. John Wiley & Sons, Ltd. 2014. Vol. 232. N. 2. P. 112–120.
-
Leppkes M. et al. RORγ-Expressing Th17 Cells Induce Murine Chronic Intestinal Inflammation via Redundant Effects of IL-17A and IL-17F // Gastroenterology. W.B. Saunders. 2009. Vol. 136. N. 1. P. 257–267.
-
Amre D.K. et al. Interleukin 10 (IL-10) gene variants and susceptibility for paediatric onset Crohn’s disease // Aliment. Pharmacol. Ther. John Wiley & Sons, Ltd. 2009. Vol. 29. N. 9. P. 1025–1031.
-
Кит О.И. и др. Воспаление и рак толстой кишки. Молекулярноиммунологические механизмы // Вопросы онклогии. 2018. Т. 64. № 1. [Kit O.I. et al. Vospalenie i rak tolstoj kishki. Molekulyarnoimmunologicheskie mexanizmy’ // Voprosy’ onklogii. 2018. Vol. 64. N. 1. (In Russ.)].
-
Zhang Y.Z., Li Y.Y. Inflammatory bowel disease: Pathogenesis // World J. Gastroenterol. Baishideng Publishing Group Inc. 2014. Vol. 20. N. 1. P. 91.
-
Loftus E.V. Clinical epidemiology of inflammatory bowel disease: incidence, prevalence, and environmental influences // Gastroenterology. W.B. Saunders. 2004. Vol. 126. N. 6. P. 1504–1517.
-
Tian T., Wang Z., Zhang J. Pathomechanisms of Oxidative Stress in Inflammatory Bowel Disease and Potential Antioxidant Therapies // Oxid. Med. Cell. Longev. Hindawi Limited. 2017. Vol. 2017.
-
Joossens M. et al. Dysbiosis of the faecal microbiota in patients with Crohn’s disease and their unaffected relatives // Gut. BMJ Publishing Group. 2011. Vol. 60. N. 5. P. 631–637.
-
Frank D.N. et al. Molecular-phylogenetic characterization of microbial community imbalances in human inflammatory bowel diseases // Proc. Natl. Acad. Sci. U.S.A. 2007. Vol. 104. N. 34. P. 13780–13785.
-
Mentella M.C. et al. Nutrition, IBD and Gut Microbiota: A Review // Nutrients. Nutrients. 2020. Vol. 12. N. 4.
-
Pickard J.M., Chervonsky A.V. Intestinal fucose as a mediator of host-microbe symbiosis // J. Immunol. 2015. Vol. 194. N. 12. P. 5588–5593.
-
Маев И.В., Андреев Д.Н. Таргетная терапия воспалительных заболеваний кишечника: реалии и перспективы // Медицинский совет. Общество с ограниченной ответственностью «Группа Ремедиум». 2018. № 6. P. 114–118. [Mayev I.V., Andreev D.N. Targetnaya terapiya vospalitel’ny’x zabolevanij kishechnika: realii i perspektivy’ // Medicinskij sovet. Obshhestvo s ogranichennoj otvetstvennost’yu “Gruppa Remedium”. 2018. N. 6. P. 114–118. (In Russ.)].
-
Князев О.В. Коррекция микрофлоры кишечника у больных язвенным колитом // Экспериментальная и клиническая гастроэнтерология. Общество с ограниченной ответственностью «Глобал Медиа технологии». 2011. № 5. [Knyazev O.V. Korrekciya mikroflory’ kishechnika u bol’ny’x yazvenny’m kolitom // E’ksperimentalnaya i klinicheskaya gastroe’nterologiya. Obshhestvo s ogranichennoj otvetstvennost’yu “Global Media texnologii”. 2011. N. 5. (In Russ.)].
-
Okamoto R., Mizutani T., Shimizu H. Development and Application of Regenerative Medicine in Inflammatory Bowel Disease // Digestion. S. Karger AG. 2023. Vol. 104. N. 1. P. 24–29.
-
Seyedian S.S., Nokhostin F., Malamir M.D. A review of the diagnosis, prevention, and treatment methods of inflammatory bowel disease // J.Med. Life. Carol Davila University Press. 2019. Vol. 12. N. 2. P. 113.
-
Фадеев Д.С. и др. Основные терапевтические мишени при болезни крона и язвенном колите // Современные проблемы науки и образования. 2022. № 5. [Fadeev D.S. et al. Osnovnye terapevticheskie misheni pri bolezni krona i yazvennom kolite // Sovremennye problemy nauki i obrazovaniya. 2022. N. 5. (In Russ.)].
-
Wallace J.L. et al. Muc-2-deficient mice display a sex-specific, COX-2-related impairment of gastric mucosal repair // Am. J. Pathol. 2011. Vol. 178. N. 3. P. 1126–1133.
-
Buisine M.P. et al. Mucin gene expression in intestinal epithelial cells in Crohn’s disease // Gut. BMJ Publishing Group. 2001. Vol. 49. N. 4. P. 544.
-
Johansson M.E.V. et al. The inner of the two Muc2 mucin-dependent mucus layers in colon is devoid of bacteria // Proc. Natl. Acad.Sci. U.S.A. National Academy of Sciences. 2008. Vol. 105. N. 39. P. 15064.
-
Ambort D. et al. Calcium and pH-dependent packing and release of the gel-forming MUC2 mucin // Proc. Natl. Acad. Sci. U.S.A. National Academy of Sciences. 2012. Vol. 109. N. 15. P. 5645.
-
Juge N. Microbial adhesins to gastrointestinal mucus // Trends Microbiol. 2012. Vol. 20. N. 1. P. 30–39.
-
Kang Y. et al. The Role and Function of Mucins and Its Relationship to Inflammatory Bowel Disease // Front. Med. Frontiers Media SA. 2022. Vol. 9. P. 848344.
-
Thomès L., Bojar D. The Role of Fucose-Containing Glycan Motifs Across Taxonomic Kingdoms // Front. Mol. Biosci. Frontiers Media S.A. 2021. Vol. 8.
-
Litvinova E.A. et al. Dietary Fucose Affects Macrophage Polarization and Reproductive Performance in Mice // Nutrients. Multidisciplinary Digital Publishing Institute (MDPI). 2021. Vol. 13. N. 3. P. 1–15.
-
Bets V.D. et al. Role of Mucin 2 Glycoprotein and L-fucose in Interaction of Immunity and Microbiome within the Experimental Model of Inflammatory Bowel Disease // Biochem. Pleiades journals. 2022. Vol. 87. N. 4. P. 301–318.
-
Larsson J.M.H. et al. Altered O-glycosylation profile of MUC2 mucin occurs in active ulcerative colitis and is associated with increased inflammation // Inflamm. Bowel Dis. Oxford Academic. 2011. Vol. 17. N. 11. P. 2299–2307.
-
Velcich A. et al. Colorectal cancer in mice genetically deficient in the mucin Muc2 // Science. 2002. Vol. 295. N. 5560. P. 1726–1729.
-
Bergstrom K.S.B. et al. Muc2 Protects against Lethal Infectious Colitis by Disassociating Pathogenic and Commensal Bacteria from the Colonic Mucosa // PLoS Pathog. Public Library of Science. 2010. Vol. 6. N. 5.
-
Van der Sluis M. et al. Muc2-deficient mice spontaneously develop colitis, indicating that MUC2 is critical for colonic protection // Gastroenterology. 2006. Vol. 131. N. 1. P. 117–129.
-
Goyal N. et al. Animal models of inflammatory bowel disease: A review // Inflammopharmacology. Birkhauser Verlag AG. 2014. Vol. 22. N. 4. P. 219–233.
-
Te Velde A.A. et al. Comparative analysis of colonic gene expression of three experimental colitis models mimicking inflammatory bowel disease // Inflamm. Bowel Dis. Oxford Academic. 2007. Vol. 13. N. 3. P. 325–330.
-
Unutmaz D., Pulendran B. The gut feeling of Treg cells: IL-10 is the silver lining during colitis // Nat. Immunol. Nature Publishing Group. 2009. Vol. 10. N. 11. P. 1141–1143.
-
Velcich A. et al. Colorectal cancer in mice genetically deficient in the mucin Muc2 // Science (80). American Association for the Advancement of Science. 2002. Vol. 295. N. 5560. P. 1726–1729.
-
Kawashima H. Roles of the gel-forming MUC2 mucin and its O-glycosylation in the protection against colitis and colorectal cancer // Biol. Pharm. Bull. 2012. Vol. 35. N. 10. P. 1637–1641.
-
Litvinova E.A. et al. Role of intestinal mucin-2 in the effectiveness of the treatment of Helicobacter spp. infection in laboratory mice // Vavilov J. Genet. Breed. Institute of Cytology and Genetics, SB RAS. 2015. Vol. 19. N. 4. P. 494.
-
Lou Z., Maher V.M., McCormick J.J. Identification of the promoter of human transcription factor Sp3 and evidence of the role of factors Sp1 and Sp3 in the expression of Sp3 protein // Gene. 2005. Vol. 351. P. 51–59.
-
Joshi S. et al. Genetically engineered mucin mouse models for inflammation and cancer // Cancer Metastasis Rev. NIH Public Access. 2015. Vol. 34. N. 4. P. 593.
-
Park Y.H. et al. Adequate Dextran Sodium Sulfate-induced Colitis Model in Mice and Effective Outcome Measurement Method // J. Cancer Prev. Korean Society of Cancer Prevention. 2015. Vol. 20. N. 4. P. 260.
-
Paassen N.B. Van et al. Colitis development during the suckling-weaning transition in mucin muc2-deficient mice // Am. J. Physiol. Gastrointest. Liver Physiol. American Physiological Society Bethesda, MD. 2011. Vol. 301. N. 4. P. 667–678.
-
Borisova M.A. et al. Mucin-2 knockout is a model of intercellular junction defects, mitochondrial damage and ATP depletion in the intestinal epithelium // Sci. Reports. Nature Publishing Group. 2020. Vol. 10. N. 1. P. 1–17.
-
Bouma G., Strober W. The immunological and genetic basis of inflammatory bowel disease // Nat. Rev. Immunol. Nature Publishing Group. 2003. Vol. 3. N. 7. P. 521–533.
-
Renes I.B. et al. Epithelial proliferation, cell death, and gene expression in experimental colitis: alterations in carbonic anhydrase I, mucin MUC2, and trefoil factor 3 expression // Int. J. Colorectal Dis. 2002. Vol. 17. N. 5. P. 317–326.
- Obermeier F. et al. CpG motifs of bacterial DNA essentially contribute to the perpetuation of chronic intestinal inflammation // Gastroenterology. 2005. Vol. 129. N. 3. P. 913–927.
Поиск



