
Биоэтические и экономические аспекты в основе выбора метода изучения токсичности лекарственных средств при однократном введении
Авдеева О.И., Макарова М.Н., Калатанова А.В., Ковалева М.А. Биоэтические и экономические аспекты в основе выбора метода изучения токсичности лекарственных средств при однократном введении. Лабораторные животные для научных исследований. 2018; 1. https://doi.org/10.29296/2618723X-2018-01-01
Резюме
Исследование общей токсичности лекарственных средств является важным аспектом их доклинического изучения, так как позволяет оценить риски при клинических исследованиях. Результаты изучения токсичности лекарственных средств при однократном введении позволяют не только обосновать дозы для исследования токсичности при многократном введении и специфических видов токсичности, но и оценить целесообразность дальнейшего исследования препарата. Проведен анализ экспериментального изучения токсичности при однократном введении воспроизведенного препарата ритонавира, в ходе анализа было доказано: исследования токсичности при однократном введении необходимо проводить на 2 видах животных, в том числе обоих полов, для исключения видовой и гендерной специфичности. Данные литературы о летальных дозах субстанций нельзя абсолютно экстраполировать на токсичность препарата в готовой лекарственной форме. При необходимости исследования токсичности в широком диапазоне доз целесообразно использовать дизайн протокола OECD 425.
Введение
Оценка токсичности при однократном введении является важной частью доклинического изучения безопасности лекарственных средств (ЛС), поскольку позволяет определить класс токсичности соединения, основные органы-мишени, прогнозировать дозы для изучения специфических видов токсичности и токсичности при многократном введении, а также соотнести терапевтические и токсические дозы. Если речь идет о воспроизведенных ЛС, результаты этого исследования позволят оценить эквитоксичность тестируемого препарата относительно референтного препарата и сделать прогноз целесообразности дальнейшего изучения тестируемого препарата.
Под изучением токсичности при однократном введении (острой токсичности) понимают токсикометрическую характеристику фармакологического вещества или лекарственного препарата, выражающую его способность вызывать гибель животных при однократном введении или при введении через короткие (не более 6 ч) интервалы времени в течение 1 сут [1]. Существует несколько вариантов изучения острой токсичности, согласно отечественным и зарубежным нормативным документам [2], все они позволяют определить переносимые, токсические и летальные дозы фармакологического вещества, причины наступления гибели животных и оценить клиническую картину интоксикации.
Объем исследования острой токсичности включает оценку следующих показателей:
- летальность;
- клиническая картина интоксикации;
- динамика массы тела;
- динамика потребления корма и воды;
- оценка поведенческих реакций;
- макроскопическое исследование внутренних органов с оценкой их массовых коэффициентов;
- микроскопические исследования (при недостаточности; макроскопических исследований для однозначного выявления органов-мишеней).
Согласно отечественным и зарубежным нормативным документам, существует ряд вариантов изучения острой токсичности. Но независимо от того, какой дизайн исследования будет использован, основанием для выбора экспериментальных доз являются:
- данные литературы о летальных дозах ЛС;
- данные литературы о летальных дозах структурных аналогов ЛС;
- прогноз о токсичности на основании химической структуры вещества.
Из всех вариантов изучения острой токсичности 2 из них позволяют точно рассчитать ЛД50. Рассмотрим эти варианты.
Вариант 1. Дизайн исследования соответствует требованиям Руководства по проведению доклинических исследований лекарственных средств [1]. Его достоинства: приемлемость использования для любых веществ и всех путей введения. К недостаткам метода можно отнести финансовую затратность и избыточное использование животных, так как требуется тестирование не менее 5 доз с обязательным достижением ЛД0 и ЛД100, т. е. в каждой группе должно быть не менее
5 самцов и 5 самок в случае мелких лабораторных животных (крысы, мыши, хомяки) и не менее 4 самцов и 4 самок в случае более крупных животных (кролики, морские свинки, карликовые свиньи).
Вариант 2. Изучение острой токсичности для токсичных веществ методом
up/down (вверх/вниз) на основании OECD 425 [3]. Данный дизайн экономически более выгоден, так как на каждый шаг затрачивается по 1 животному каждого пола. При правильном подборе стартовой дозы для исследования может потребоваться только по 6 животных каждого пола, в случае неблагоприятного выбора стартовой дозы – по 18 животных каждого пола.
Если тестируемый препарат на основании данных литературы малотоксичен, то для оценки токсичности при однократном введении может быть использован протокол OECD 420 [4]. При этом вводят только 1 дозу препарата в 2 этапа, сначала 1 самцу и 1 самке, затем – еще 4 самцам и 4 самкам. Выбор экспериментальной дозы определяется следующими исходными условиями исследования:
- Экспериментатор располагает достоверной информацией о том, что исследуемое вещество нетоксично: тестируемая доза – 2000 мг/кг (лимит согласно отечественным и зарубежным стандартам) [1, 4].
- На основании имеющихся данных вещества могут быть отнесены к группе малотоксичных веществ: 2000 мг/кг < ЛД50 < 5000 мг/кг. В исключительных случаях, когда это действительно необходимо, может быть протестирована доза 5000 мг/кг. Следуя гуманным принципам обращения с животными, тестирования дозы 5000 мг/кг следует избегать и проводить его только тогда, когда полученные данные действительно внесут существенный вклад в изучение безопасности данного вещества (для людей, животных, окружающей среды).
- Максимальная доза ограничена предельными объемами для введения. Исследование направлено на изучение сравнительной токсичности лекарственных форм, применение которых заведомо не позволит достичь максимальных доз, приемлемых для изучения острой токсичности. В этом случае определяется переносимость предельной для введения лабораторным животным дозы.
Таким образом, важное отличие изучения острой токсичности препаратов, согласно протоколам OECD, – бережное отношение к лабораторным животным, использование их только в минимальных количествах, необходимых для оценки действия препарата. Использование минимального количества животных приводит к существенному снижению стоимости исследований и, как следствие, указывает на целесообразность его проведения.
Кроме того, важно учитывать интервал между введениями различных доз, что обусловлено характером течения интоксикации. Введение следующей дозы следует начинать только в случае, если есть уверенность в том, что все животные на текущей дозе выживут. Рекомендуется интервал в 3–4 дня между тестированиями, что также обеспечивает экономное расходование лабораторных животных.
Руководствуясь биоэтическими принципами обращения с лабораторными животными, целесообразно применять различные методы изучения острой токсичности, не уступающие друг другу по информативности. Выбор метода зависит от имеющихся данных о лекарственном веществе.
Для иллюстрации некоторых сложностей «простого» метода изучения токсичности при однократном введении приведен анализ результатов сравнительного изучения острой токсичности препаратов ритонавира при внутрижелудочном введении на 2 видах лабораторных животных (крысах и мышах) с анализом влияния видовой специфичности на токсические эффекты.
Разбор экспериментального случая
В ходе проведенного исследования была изучена острая токсичность воспроизведенного препарата ритонавира для перорального применения [Т] в сравнении с референтным препаратом – препаратом ритонавира для перорального применения, зарегистрированным в РФ [R]. В качестве тест-системы были использованы половозрелые аутбредные крысы массой тела 200–300 г в возрасте 10–12 нед и половозрелые аутбредные мыши массой тела 25–35 г в возрасте 7–9 нед. Животные получены в НПО «ДОМ ФАРМАЦИИ». На момент начала исследования животные были здоровы (получены данные клинического осмотра и серологических тестов). Адаптация к условиям эксперимента составила 5 дней. Препараты вводили животным внутрижелудочно (как аналог перорального применения в клинике) и через зонд в виде суспензии. В соответствии с техническим заданием спонсора дизайн исследования был выбран согласно требованиям Руководства по проведению
доклинических исследований лекарственных средств [1]. В каждой группе крыс было по 5 самцов и 5 самок, мышей – по 7 самцов и 7 самок. Животные содержались в стандартных условиях вивария. Исследование выполнено в соответствии с принципами GLP и Европейской конвенции по защите позвоночных животных (одобрено на заседании биоэтического комитета; заключение №БЭК 4.46/16).
С целью оценки острой токсичности препараты вводили однократно, регистрацию летальных эффектов осуществляли в течение 14 дней, расчет величин летальных доз проводили методом наименьших квадратов для пробит-анализа кривых летальности [5].
Выбор доз осуществляли на основании имеющихся данных литературы о токсических дозах ритонавира:
- минимальная летальная доза составила 20 × высших терапевтических доз (ВТД) – для крыс (≈2000 мг/кг) и 10 × ВТД – для мышей (≈2000 мг/кг) [6];
- ЛД50 при внутрижелудочном введении крысам и мышам >2500 мг/кг [7, 8];
- ЛД50 при внутрижелудочном введении мышам – 1030–1970 мг/кг [9];
- ритонавир отнесен к 4-й категории токсичности при внутрижелудочном введении, т.е. 300 < ЛД50 < 2000 мг/кг [10].
Таким образом, для изучения острой токсичности на крысах были протестированы дозы 250–2500 мг/кг, на мышах – 148–2500 мг/кг для обоих препаратов.
Зависимые от доз летальные эффекты исследуемых объектов для крыс представлены в табл. 1.
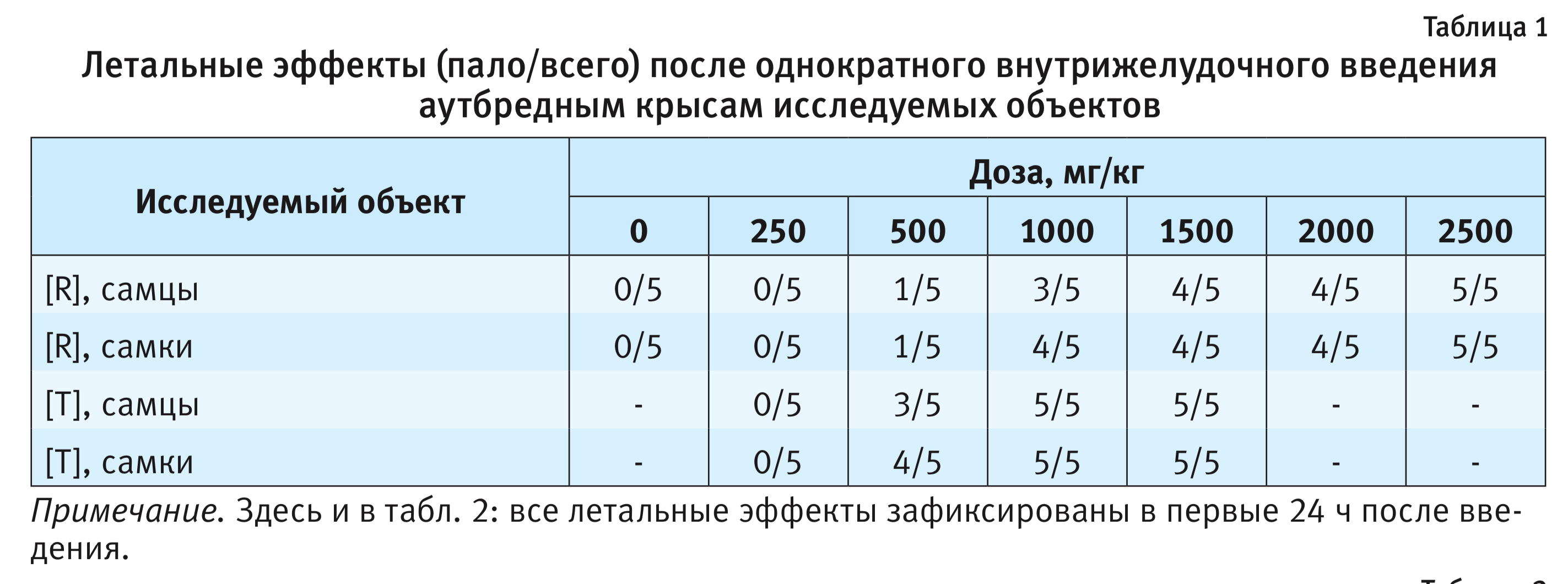
Результаты токсикометрии поставили под сомнение гипотезу об эквитоксичности исследуемых препаратов. Для исключения видовой специфичности аналогичное исследование было проведено на мышах.
Зависимые от доз летальные эффекты исследуемых объектов для мышей представлены в табл. 2.
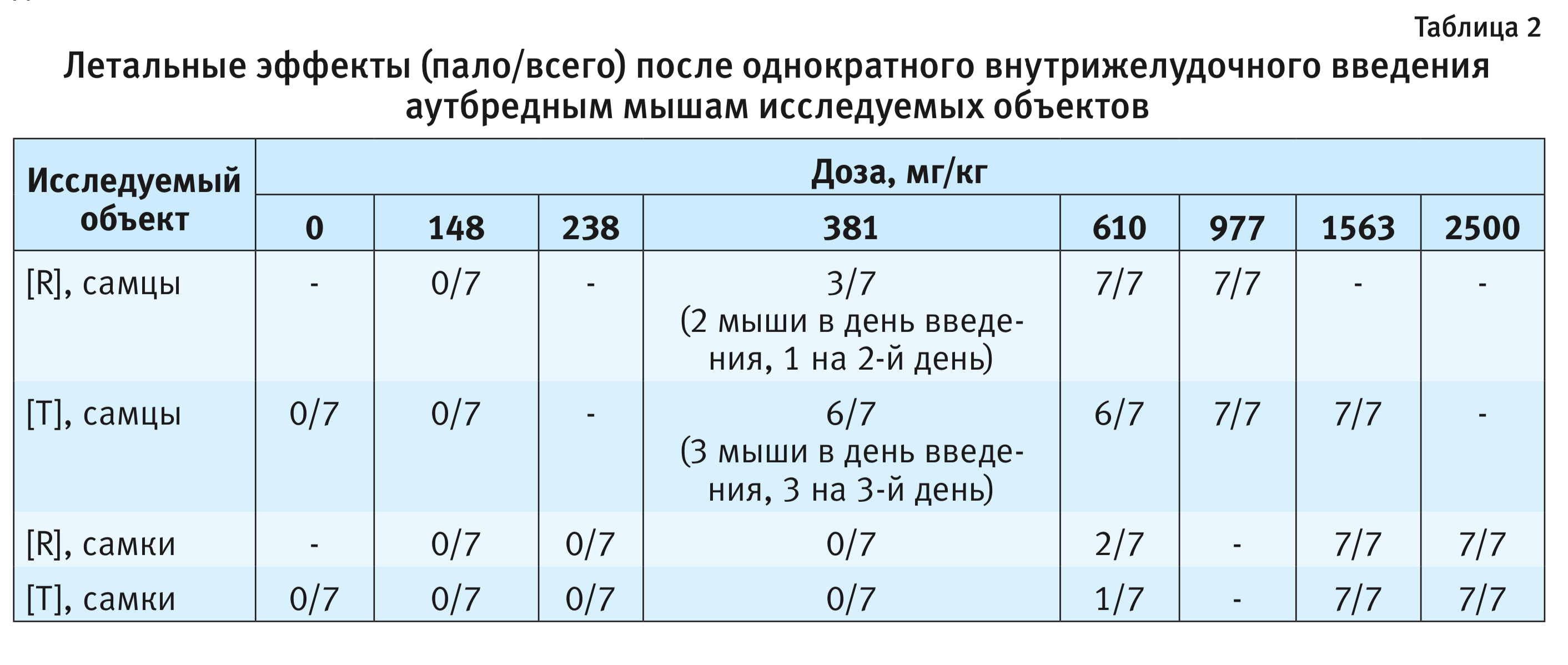
Установленная в ходе проведения эксперимента гибель крыс и мышей позволила оценить токсичность исследуемых объектов, результаты расчетов ЛД50 представлены в табл. 3.
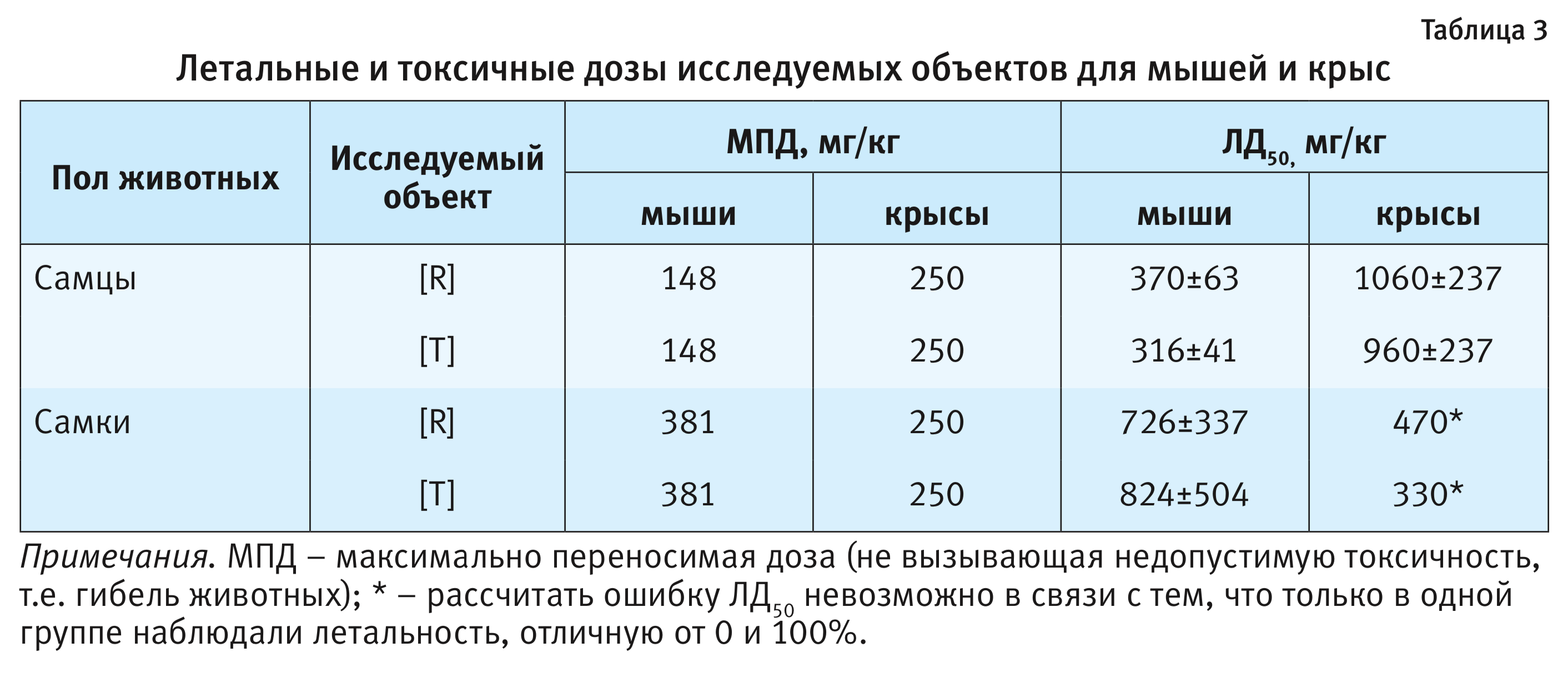
Исследование сравнительной острой токсичности исследуемых препаратов на крысах и мышах выявило 2 важных момента – видовую и половую специфичность токсического действия препаратов. Следовательно, целесообразно изучение острой токсичности на 2 видах животных не только для оригинальных, но и для воспроизводимых препаратов. В ходе исследования однозначно подтверждена необходимость проведения исследований токсичности на животных обоих полов, так как в противном случае могут быть не зарегистрированы некоторые нежелательные эффекты, которые потом будут выявлены при клинических исследованиях, что категорически недопустимо.
Кроме того, экспериментально установленные летальные дозы существенно отличаются от данных, представленных в литературе, что потребовало для оценки параметров токсичности участия недопустимого с точки зрения биоэтических принципов количества животных: 110 крыс и 161 мыши. При использовании метода «вверх/вниз», согласно протоколу OECD 425, та же цель была бы достигнута
с участием 18 животных каждого пола при тестировании 1000-кратного диапазона доз (например, 1,75; 5,5; 17,5; 55; 175; 550; 2000 мг/кг).
Выявленное несоответствие вполне закономерно, так как большинство сведений о безопасности вещества дано для субстанции, а в комбинации со вспомогательными веществами лекарственное соединение способно изменить свои
фармакокинетические характеристики и, как следствие, токсические.
Заключение
На основании проведенных исследований можно сделать следующие выводы:
- выбор доз на основании данных литературы не гарантирует адекватность протестированного диапазона доз, так как большинство сведений представлено для субстанций, а не для лекарственных форм;
- использование метода «вверх/вниз» даже при неудачном выборе стартовой дозы (при несоответствии экспериментальной ЛД50 данным литературы) позволяет протестировать широкий диапазон доз с использованием минимального количества животных;
- при изучении острой токсичности даже воспроизведенных препаратов целесообразно использовать не менее 2 видов лабораторных животных для исключения особенностей видовой токсичности;
- показана необходимость участия в эксперименте животных обоего пола в отличие от требований ряда протоколов OECD, рекомендующих использовать только 1 пол;
- изучение острой токсичности воспроизведенных препаратов целесообразно, так как позволяет не только скорректировать дозы при анализе токсичности в случае многократного введения, но и выбрать вид животных, а также оценить в целом перспективность дальнейшего изучения воспроизведенного препарата;
- оценка общей токсичности воспроизведенных препаратов, в том числе при однократном введении, необходима для обеспечения национальной фармацевтической безопасности, так как вспомогательные вещества, входящие в состав генериков, отличаются количественно и качественно от зарегистрированного аналога и по-другому влияют на профиль токсичности готовых лекарственных средств.
Список источников
- Руководство по проведению доклинических исследований лекарственных средств. Часть первая. М.: Гриф и К, 2012; 944.
- Авдеева О.И., Макаренко И.Е., Макарова М.Н., Шекунова Е.В., Кашкин В.А., Макаров В.Г. Гармонизация исследований по проведению острой токсичности в соответствии с российскими и зарубежными требованиями. Международный вестник ветеринарии. 2015; 1: 103–109.
- OECD (2008) Guideline for testing of chemicals. Acute Oral Toxicity – Up-and-Down-Procedure (UDP)
- № 425.
- OECD (2001) Guideline for testing of chemicals. Acute Oral Toxicity – Fixed Dose Procedure № 420.
- Прозоровский В.Б. Практическое пособие по ускоренному определению средних эффективных доз и концентраций биологически активных веществ. Байкальск: Общество духовной и психической культуры. 1994: 46.
- NORVIR® (ritonavir capsules) Soft Gelatin (ritonavir oral solution). FDA’s 8/29/08 approved draft PI. 46.
- Norvir®: Ritonavir film-coated tablets (100 mg) [product monograph]. Сontrol No: 201368, AbbVie Corporation 2017. Электронный ресурс [http://www.abbvie.ca/content/dam/abbviecorp/ca/english/docs/NORVIR_PM_EN.pdf]
- MSDS и ЛД50 Ритонавир. Электронный ресурс: https://s3-us-west-2.amazonaws.com/drugbank/msds/DB00503.pdf?1497888312
- MSDS и ЛД50 Ритонавир. Электронный ресурс: https://www.cymitquimica.com/uploads/products/45/pdf/sds-1604803.pdf
- MSDS и ЛД50 Ритонавир. Электронный ресурс: http://194.7.155.215/DirectWebViewer/private/document.aspx?prd=ACR46122~~PDF~~MTR~~CLP1~~EN~~2016-03-17%2003:38:50~~Ritonavir~~
Поиск



