Генетически модифицированные линии лабораторных животных, используемые в качестве модели метаболического синдрома и сахарного диабета
Ковалева М.А., Макарова М.Н., Макаров В.Г. Генетически модифицированные линии лабораторных животных, используемые в качестве модели метаболического синдрома и сахарного диабета. Лабораторные животные для научных исследований. 2018; 1. https://doi.org/10.29296/2618723X-2018-01-03
Резюме
Распространенность сахарного диабета и метаболического синдрома в современном обществе ежегодно увеличивается, что становится причиной повышенной заболеваемости и смертности от отсроченных осложнений. Сегодня метаболические патологии являются социально значимыми заболеваниями, ведущими к резкому ухудшению качества жизни пациента. Использование адекватных экспериментальных моделей позволяет понять причины и механизмы развития сахарного диабета и метаболического синдрома, исследовать потенциальные методы их профилактики и терапии. Метаболический синдром представляет собой многофакторный симптомокомплекс патологических изменений, и выбор экспериментальной модели является основополагающим для получения объективных результатов. Представлены наиболее надежные и изученные генетические модели сахарного диабета и метаболического синдрома, систематизированы виды лабораторных животных. Приведены основные характеристики линий мышей и крыс, в том числе биохимические, характеризующие состояние углеводного (концентрация глюкозы и инсулина), липидного обмена (концентрации холестерина и триглицеридов), энергетического обмена (лептин). Описаны линия мышей с дефицитом лептина Lepob/ob, линии крыс и мышей с дефицитом рецепторов лептина LepRdb/db, Zucker fatty rats, Zucker Diabetic Fatty rats, подштаммов Spontaneoulsy hypertensive rats, Obese spontaneously hypertensive rats/Koletsky rats и SHR/NDmc-corpulent rats. Рассмотрены также трансгенные мыши МС4R-/-, Agouti yellow, KKAy/a, для которых характерно развитие резистентности к лептину за счет дефекта передачи сигнала лептина в клетку. Для крыс также приведены биометрические параметры артериального давления. Приводится информация об изменениях основных характеристик в зависимости от пола и возраста лабораторных животных. Каждая из рассмотренных линий грызунов имеет специфические свойства, характерные для сахарного диабета и метаболического синдрома, что позволяет всесторонне изучать механизмы развития патологий, а также потенциальных лекарственных препаратов для терапии данных заболеваний.
Введение
В настоящее время проблема распространения патологий, связанных с метаболическими нарушениями, такими как сахарный диабет (СД), ожирение и ассоциированный с ним метаболический синдром (МС), является крайне актуальной. По прогнозам Всемирной организации здравоохранения (ВОЗ), к 2025 г. ожидается рост числа пациентов с метаболическими нарушениями в среднем на 50%. Эта тенденция требует более углубленного изучения патологических процессов на молекулярном уровне, разработку и внедрение новых лекарственных препаратов, что требует использования адекватных экспериментальных моделей, в том числе с использованием генетически модифицированных животных.
Создание и использование генетически модифицированных животных, как моделей метаболических нарушений, продолжает оставаться весьма перспективным направлением для изучения молекулярных механизмов развития патологий и оценки фармакологической активности тестируемых объектов. Следует отметить, что трансгенные животные, как правило, являются моногенными, т.е. патологические изменения связаны с дефектом функции только одного белка. Генетические модели заболеваний получают либо путем селекции животных со спонтанными мутациями, которые закреплены в череде поколений, либо искусственно индуцируют утрату заданного гена. В данном обзоре литературы рассмотрены линии лабораторных животных, имеющие спонтанные или индуцированные мутации, используемые для моделирования сахарного диабета и метаболического синдрома. В литературных источниках, опубликованных на русском языке, недостаточно информации о трансгенных линиях грызунов, используемых для моделирования СД и МС. В данной работе предпринята попытка привести основные характеристики трансгенных линий грызунов.
Трансгенные линии крыс
В научной практике наиболее известной животной моногенной моделью для изучения метаболического синдрома являются крысы линий fa/fa Zucker fatty rats (ZFR) и Zucker diabetic fatty rat (ZDF) [1].
Линия животных ZFR имеет мутацию гена рецептора лептина на 5-й хромосоме. Данный дефект приводит к снижению
связывания лептина с поверхностью рецептор-экспрессирующих клеток, при этом сродство к лептину не
изменяется, далее происходит развитие устойчивости к лептину в головном мозге. Следствием описанных
патологических изменений является развитие гиперфагии и ожирения к 4-й неделе жизни лабораторных животных.
Эти нарушения сочетаются с незначительной гипергликемией, резистентностью к инсулину, гиперинсулинемией,
гиперлипидемией и умеренной гипертензией. К 14-й неделе жизни количество депонированных липидов
составляет около 40% по отношению к массе тела. У животных развивается гиперплазия
и гипертрофия адипоцитов. Островки Лангерганса гипертрофированы, их количество увеличено. Кроме того,
у ZFR после 14-й недели жизни установлено развитие нефропатии. В плазме крови повышено содержание
холестерина (ХС), жирных кислот и триглицеридов (ТГ), а в печени обнаружена избыточная продукция
липопротеинов. Увеличение концентрации триглицеридов в крови связано с накоплением липопротеинов очень
низкой плотности (ЛПОНП). Увеличение уровня холестерина в крови связано с его увеличением во фракциях
ЛПОНП и липопротеинов высокой плотности (ЛПВП) [2]. Следует отметить, что ожирение у данной линии
животных, наряду с резистентностью рецепторов головного мозга к лептину, связано с состоянием
хронического воспаления, характеризующегося аномальным образованием провоспалительных медиаторов [3], включая фактор
некроза опухоли-α (ФНО-α) и NO-синтазу, а также дефицит энергии в виде АТФ.
А.
Picchi и соавт. полагают, что именно избыточная экспрессия ФНО-α приводит у ZFR к развитию
эндотелиальной дисфункции на фоне индукции НАДФ-оксидазы с последующим образованием супероксид-анионов [4].
Указанная линия животных может быть использована как самостоятельная модель МС, патологические изменения
у животных развиваются при содержании на стандартном рационе.
Крысы линии Zucker diabetic fatty rat (ZDF) избирательно инбредные к гипергликемии, являются подштаммом линии ZFR. ZDF несут аутосомно-рецессивный дефект транскрипции β-клеток поджелудочной железы, наследование которого происходит независимо от мутации гена рецептора лептина (Lepr). Следует отметить, что ген, ответственный за дефект, до сих пор не определен. Установлено, что этого дефекта недостаточно, чтобы вызвать СД, и только в сочетании с мутацией гена Lepr может развиваться гипергликемия [5]. ZDF в меньшей степени страдают ожирением, но больше инсулинорезистентны, чем крысы ZFR. Самцы склонны к развитию СД, который формируется к 7–10-й неделе жизни лабораторных животных. У женских особей развиваются ожирение, резистентность к инсулину, но без развития СД. У Zucker diabetic fatty rat гипергликемия, гиперинсулинемия и гипертриглицеридемия формируются к 12–15-й неделе жизни наряду с изменение диастолического и систолического артериального давления (САД) (табл. 1). В литературных источниках встречаются противоречивые данные о сроках развития гипергликемии у ZDF и о концентрации глюкозы в периферической крови. Есть данные, что к 10–15-й неделе жизни концентрация глюкозы в периферической крови животных может достигать 500 мг/дл (28 ммоль/л) [6, 7].
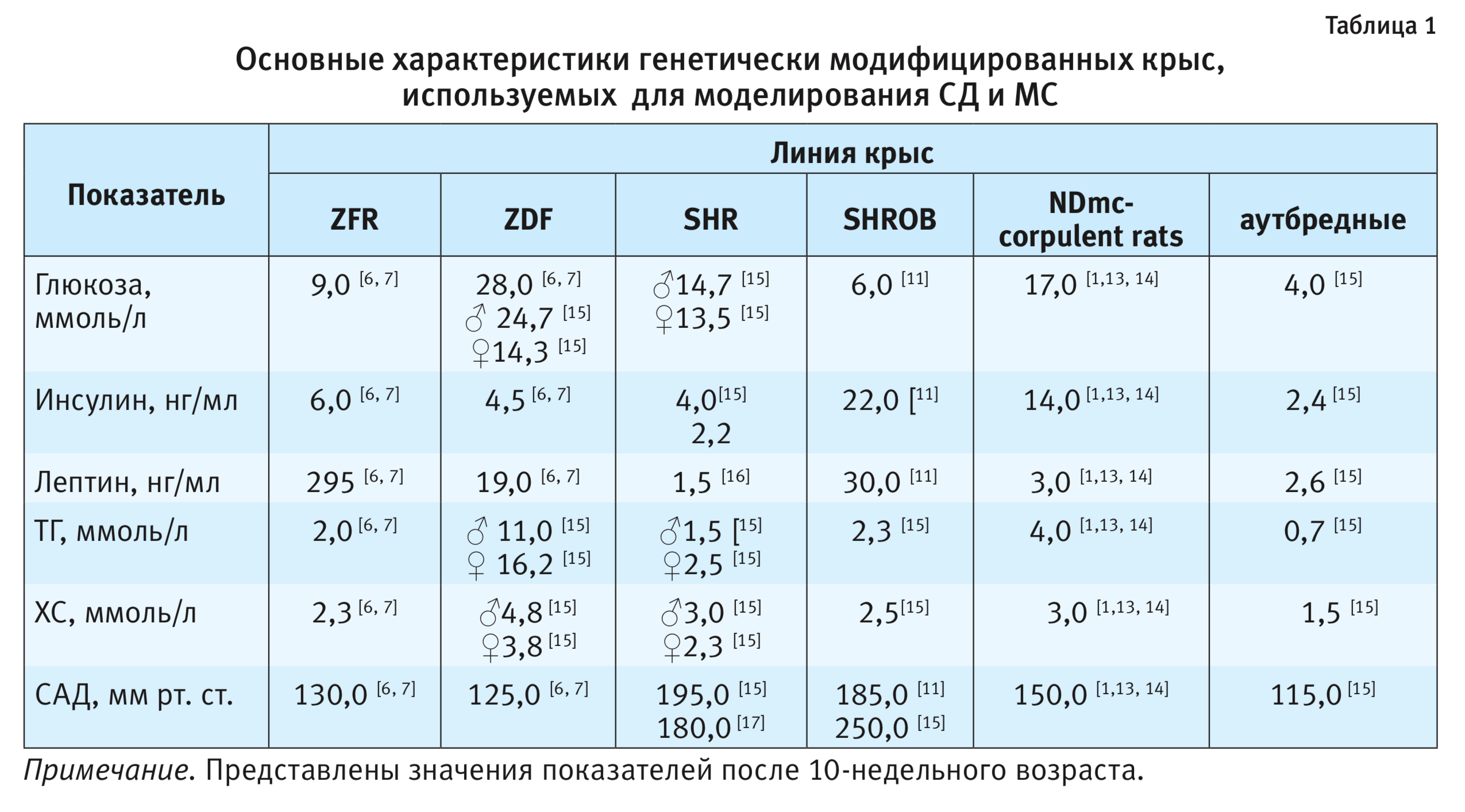
J.B. Clark и соавт. приводит данные о развитии гипергликемии лишь в возрасте 6 мес [8]. Параллельно у этой линии животных наблюдают умеренное увеличение САД. В возрасте 20 нед содержание холестерина в сыворотке крови у ZDF в 2,5 раза выше по сравнению с Leprfa штаммом. Альбуминурия в возрасте 31-й недели сопровождается утолщением базальной мембраны и клубочковым фиброзом (после 47-й недели жизни). Увеличение триглицеридов в печени регистрируется к 20-й неделе жизни. Исследования β-клеток ZDF показали, что основной дефект состоит в увеличении скорости их апоптоза. Снижение синтеза инсулина и подавление функции транспортера глюкозы ГЛЮТ-2 неизбежно приводит к развитию гипергликемии. Снижение транспорта глюкозы также связано с уменьшением уровня ГЛЮТ-4 в жировой ткани и скелетных мышцах ZDF [9]. ZDF могут быть использованы как самостоятельная модель изучения метаболических нарушений, однако для воспроизведения более полной картины метаболического синдрома рекомендовано содержать самок крыс на высокожировой диете с содержанием жиров более 48% от суточной калорийности. Самцы становятся тучными и демонстрируют гипергликемию даже при содержании на стандартном рационе вивария [10].
Спонтанно гипертензивные крысы (Spontaneoulsy hypertensive rats, SHR) – известная экспериментальная модель для изучения в первую очередь гипертензии, которая также может быть использована для моделирования метаболических нарушений, в частности МС. У данных крыс развивается гипертензия, абдоминальное ожирение, гипертриглицеридемия. Как правило, данную линию содержат на высококалорийной диете с целью развития более полного и выраженного симптомокомплекса МС. На основе линии SHR выведены следующие линии крыс: спонтанно гипертензивные крысы с ожирением (Obese spontaneously hypertensive rats/Koletsky rats, SHROB), SHR/NDmc – тучные крысы (SHR/NDmc corpulent rats), которых считают как более предпочтительными моделями для создания МС, чем SHR, поскольку они имеют дефект гена рецептора лептина [1]. Гипертензивные крысы с ожирением (SHROB) имеют фенотипические признаки, характерные для МС. У данных крыс развивается гипертония, гиперинсулинемия, гиперлипидемия и нефропатия. У крыс SHROB так же, как и у крыс ZDF, имеется мутация гена рецептора лептина fak, приводящая к нарушению передачи сигнала через рецептор. Патологические изменения развиваются с 5-недельного возраста и достигают наибольшей выраженности к 30-й неделе. Уровень циркулирующего лептина у данной линии увеличивается примерно в 30 раз, вследствие чего возникают гиперфагия и увеличение массы тела. У крыс развивается гиперлипидемия даже при содержании на стандартном рационе вивария, характеризующаяся заметным увеличением концентрации триглицеридов, умеренным повышением уровня холестерина в плазме крови. Гиперинсулинемия SHROB сочетается с нормальным или умеренно повышенным уровнем глюкозы в периферической крови. Спонтанная гипертензия начинает развиваться с 3-месячного возраста и в среднем составляет 180 мм рт. ст. [11]. Сообщается, что к 30-й неделе жизни животных артериальное давление поднимается до 200–250 мм рт. ст. (см. табл. 1). У SHROB также обнаружены патологические изменения сосудов по типу атеросклероза сосудов человека. Данные изменения особенно выражены в артерии брюшной полости [12]. Крысы линии NDmc-corpulent rats также могут быть использованы в качестве животной модели метаболических нарушений. Этот инбредный подштамм демонстрирует развитие таких метаболических изменений, как гиперфагия, увеличение массы тела и жировой ткани, что сопровождается гипертонией, гипертрофией сердца, сахарным диабетом и гиперлипидемией [1, 13]. Концентрация глюкозы в периферической крови у данной линии может достигать 300 мг/дл (17 ммоль/л) при концентрации инсулина 14 нг/мл [14].
Следует отметить, что в различных публикациях значения биохимических показателей для крыс отличаются, что существенно зависит от возраста лабораторных животных, а также от их пола [15].
Трансгенные линии мышей
В качестве генетических моделей метаболических нарушений достаточно часто используют мышей Lep ob/ob и Lep db/db.
Линии мышей Lepob/ob (C57BL/6J-ob/ob) имеют моногенные аутосомно-рецессивные мутации в гене лептина на 6-й хромосоме. Данная линия была открыта в 1949 г., в лаборатории Джексона [18]. У этих животных развивается ожирение, гиперинсулинемия и гипергликемия к 4-й неделе жизни. К 11-й неделе жизни масса тела животных может достигать 60 г. Нарушение толерантности к глюкозе было зарегистрировано к 12-й неделе жизни животных. Концентрация глюкозы составляет около 300 мг/дл (17 ммоль/л), при этом средние значения концентрации инсулина составляют 40 нг/мл (табл. 2). У животных дополнительно развивается гипертрофия левого желудочка со снижением функции сердца, которая регистрируется в возрасте 24 нед, фиброз сердца установлен после 20-недельного возраста, а стеатоз печени обнаружен уже у особей в возрасте 12 нед. В отличие от клинических случаев у людей с метаболическим синдромом, описанная линия мышей демонстрирует снижение артериального давления и отсутствие дислипидемии [19, 20].
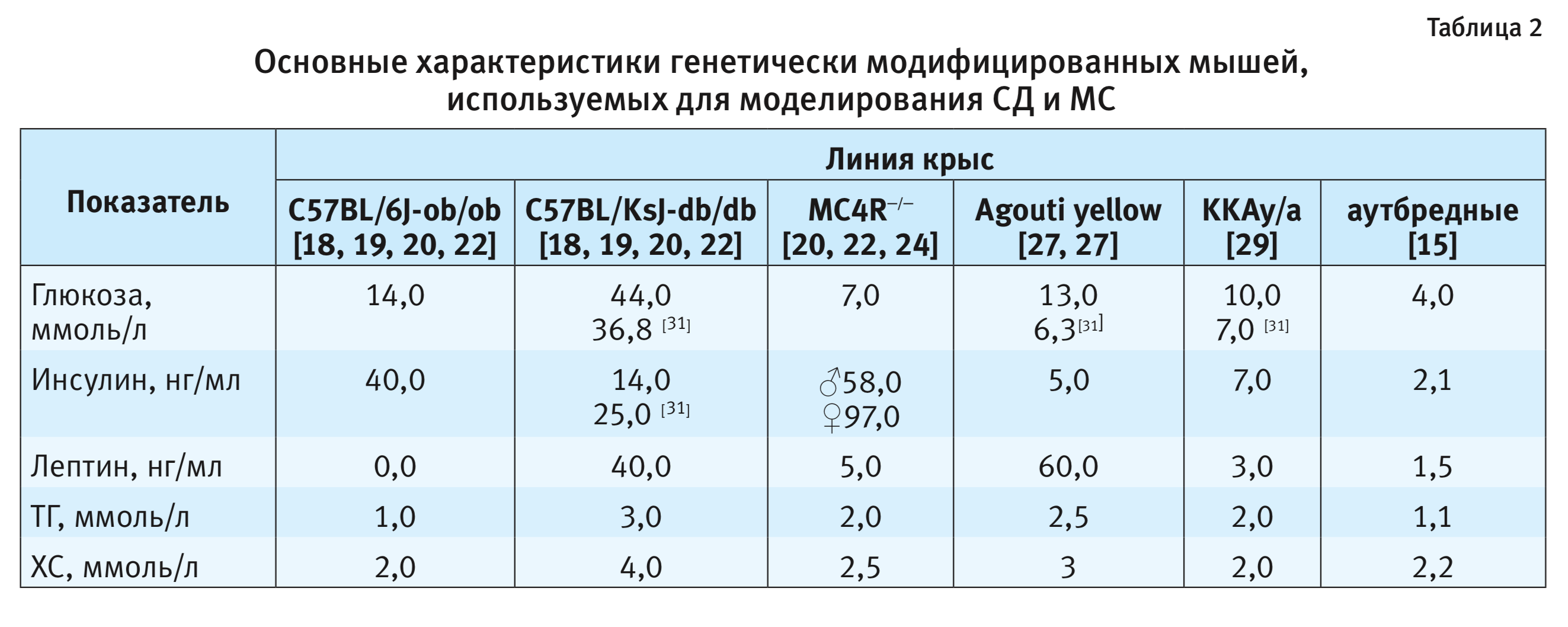
Мыши линии LepRdb/db (C57BL/KsJ-db/db) унаследовали аутосомно-рецессивную мутацию
в гене рецептора лептина, присутствующего на 4-й хромосоме [21]. Масса тела у этих животных повышается
после 6-недельного возраста и к 11-й неделе жизни превышает 55 г. Концентрация глюкозы в крови
натощак увеличивается после 8-й недели жизни и к 11-недельному возрасту составляет около 800 мг/дл
(44 ммоль/л). Увеличение содержания триглицеридов, общего холестерина и неэтерифицированных жирных кислот
в плазме крови происходит сочетанно с уменьшением соотношения ЛПВП/ЛПНП старше 13-недельного возраста.
Гиперинсулинемия и нарушение толерантности к глюкозе наблюдают после 12-недельного возраста. В этом
же возрасте в сердце развиваются как инфильтрация с воспалением, так и фиброз. Отмечается дисфункция
сосудистого эндотелия, при этом изменений артериального давления не выявлено. Стеатоз печени наблюдается после
20-недельного возраста. Метаболические профили мышей линий Lepob/ob
и LepRdb/db почти идентичны. Обе линии лабораторных животных характеризуются наличием
ожирения, гиперинсулинемии, гипергликемии, а также повышенным общим уровнем холестерина. Кроме того,
в обеих моделях отмечались нарушения размножения (бесплодие) и гормонального статуса. Основное различие
между двумя линиями лабораторных животных заключается в том, что у мышей LepRdb/db
повышена концентрация лептина циркулирующего в крови, которая пропорциональна степени ожирения, в то время
как у Lepob/ob мышей отсутствует в крови циркулирующий лептин. Поэтому
LepRdb/db модель может быть использована для выяснения влияния лептина на различные типы клеток
[19, 22].
Резистентность к лептину может развиваться и в результате дефекта передачи сигнала лептина в клетку, связанного с действием различных посредников. Центральная система меланокортина является посредником многих биологических эффектов лептина, играет важную роль в регуляции энергетического обмена и поддержании гомеостаза [23]. Механизм депонирования триглицеридов в печени опосредован описанной системой. Меланокортиновый рецептор 4-го типа (МС4R) экспрессируется в ряде ядер мозга грызунов, которые связаны с нейроэндокринными путями. Изменения в гене рецептора MC4R – наиболее распространенная моногенная причина ожирения, которая известна и характерна для людей. У мышей МС4R−/− (МС4R-дефицитные мыши) с дефектом в МС4R отмечаются многие из тех же фенотипических характеристик, что и у человека. МС4R−/− мыши имеют синдром ожирения, развивающийся на фоне гиперфагии. У животных регистрируют гипергликемию, гиперинсулинемию, увеличение мышечной массы и линейного роста, снижение величины основного обмена. Гиперинсулинемию у данных животных нельзя считать абсолютно зависимой от ожирения, поскольку у молодых МС4R−/− мышей наблюдается повышенная концентрация инсулина в периферической крови перед началом ожирения. Несмотря на выраженное ожирение в зрелом возрасте, МС4R−/− мыши не страдают гипертонией. Другой примечательной особенностью МС4R−/− дефицитных мышей является их повышенная чувствительность к высокому содержанию жиров в корме, что усугубляет гиперфагию, ожирение и гиперинсулинемию. Следует отметить, что данная линия животных недостаточно изучена с точки зрения развития дислипидемии. В частности, нет информации об изменении содержания триглицеридов и неэтерифицированных жирных кислот в плазме крови, но при этом отмечается стеатоз печени. Концентрация лептина у МС4R−/− мышей в возрасте 4–8 нед в крови повышена в 4,5 раза у самцов и 1,5 раза – у самок по сравнению с аутбредными мышами. К возрасту 17–23 нед у самок концентрация лептина достигает примерно 97 нг/мл, у самцов – около 58 нг/мл, что в 6,5 и 2,5 раза выше, чем у аутбредных мышей [24].
Мыши линии Agouti yellow (Ау/а-мыши) имеют несколько спонтанных мутаций, влияющих на экспрессию белка агути, транскрибирующегося агути-геном (А). Белок агути в норме у грызунов экспрессируется только в меланоцитах и контролирует окраску шерсти. Доминантная мутация Agouti yellow в локусе агути (хромосоме 2) вызывает повсеместную эктопическую экспрессию белка Агути [25]. Этот белок действует как антагонист меланокортинового сигнального пути, который реализует действие лептина. Такие мыши показывают разную окраску шерсти, возрастное ожирение и инсулинорезистентность за счет гиперфагии и снижения двигательной активности. Гиперинсулинемия развивается у животных к 6-й неделе жизни. Гиперплазия β-клеток поджелудочной железы наблюдается с 21-го дня жизни. У особей с ожирением отсутствуют признаки атеросклеротического поражения сосудов [26]. Agouti yellow могут воспроизводить потомство лишь до 4-месячного возраста, в отличие от нерепродуктивных мышей линий Lepob/ob и LepRdb/db.
Еще одной моделью изучения метаболических патологий являются мыши KKAy/a, которые были выведены на основе линии Kuo Kondo (КК-мыши) путем введения гена ожирения Agouti yellow в штамм [27]. У мышей KKAy/a присутствует ген ожирения и диабетический ген, в отличие от КК-мышей, у которых есть только диабетический ген. У данной модели развивается ожирение в зрелом возрасте, более серьезная гиперинсулинемия и более заметные изменения в панкреатических островках, чем у КК-мышей. Однако гиперинсулинемия и гипергликемия, наблюдаются в возрасте 6–8 нед. Причина этих изменений – эктопическая экспрессия агути-антагонистического белка-рецептора MCR4 в гипоталамусе. Считается, что инсулинорезистентность вызвана такими физиологическими изменениями, как снижение уровней адипонектина в сыворотке крови, высокая активность ферментов глюконеогенеза в печени и повышенное продуцирование ФНО-α и/или других цитокинов [28]. Начиная с 5-й недели жизни в поджелудочной железе мышей KKAy/a наблюдаются патологические изменения: панкреатические островки гипертрофированы, а β-клетки дегранулированы, что приводит к резистентности к инсулину. Кроме того для данной линии установлены патологические изменения в почках. К 10-й неделе жизни у животных развивается диффузный гломерулосклероз. Описанная модель подходит для изучения лекарственных препаратов с антидиабетической активностью, имеющих гипогликемическое действие, снижающих резистентность к инсулину или повышающих чувствительность к нему [29].
Следует отметить, что в публикациях последних 20 лет значения биохимических показателей для мышей отличаются, что существенно зависит от возраста лабораторных животных, а также от их пола [20, 22, 24, 30, 31].
Заключение
В данной статье приведен обзор данных литературы о трансгенных линиях мышей и крыс. На сегодняшний день для проведения доклинических исследований метаболических нарушений создано множество линий лабораторных животных имеющих инсулинорезистентность, гипергликемию, дислипидемию, ожирение, гипертензию. Такое биологическое разнообразие позволяет воспроизвести требуемый симптомокомплекс, характерный для СД и/или МС, всесторонне изучить не только патологические механизмы заболевания, но и фармакологическую активность тестируемых объектов. Выбор экспериментальной модели и тест-системы – неотъемлемая часть для получения достоверных результатов, оцениваемых при экспертизе регистрационного досье с целью получения разрешения на проведение клинических исследований.
Список источников
- de Artinano A.A., Castro M.M. Experimental rat models to study the metabolic syndrome. British Journal of Nutrition. 2009. Vol. 102: 1246–53.
- Zucker T.F., Zucker L.M. Hereditary obesity in the rat associated with high serum fat and cholesterol. Society for experimental biologyand medicine. 1962. Vol. 110: 165–71.
- Ouchi N., Kihara S., Funahashi T., Matsuzawa Y., Walsh K. Obesity, adiponectin and vascular inflammatory disease. Current Opinion in Lipidology. 2003. Vol. 14: 561–6.
- Picchi A., Gao X., Belmadani S., Potter B.J., Focardi M., Chilian W.M., Zhang C. Tumor necrosis factor-alpha induces endothelial dysfunction in the prediabetic metabolic syndrome. Circulation Research. 2006. Vol. 99: 69–77.
- Wang B., Chandrasekera C., Pippin J.J. Leptin- and Leptin Receptor-Deficient Rodent Models: Relevance for Human Type 2 Diabetes. Current Diabetes Reviews. 2014. Vol. 10: 131–45.
- Simmons R.K., Alberti K.G., Gale E.A. et al. The metabolic syndrome: useful concept or clinical tool? Report of a WHO expert consultation. Diabetologia. 2010. Vol. 53 (4): 600–5.
- Janssen S.W.J., Martens G.J.M., (Fred) Sweep C.G.J., Ross H.A., Hermus A.R.M.M. In Zucker Diabetic Fatty Rats Plasma Leptin Levels are Correlated with Plasma Insulin Levels rather than with Body Weight. Hormone and Metabolic Research. 1999. Vol. 31: 610–5.
- Clark J.B., Palmer C.J., Shaw W.N. The diabetic Zucker fatty rat. Society for Experimental Biologyand Medicine. 1983. Vol. 173 (1): 68–75.
- Srinivasan K., Ramarao P. Animal models in type 2 diabetes research: An overview. Indian journal of medical research. 2007. Vol. 125: 451–72.
- Лещенко Д.В., Костюк Н.В., Белякова М.Б., Егорова Е.Н., Миняев М.В., Петрова М.Б. Диетически индуцированные животные модели метаболического синдрома (обзор литературы). Верхневолжский медецинский журнал. 2015. Т. 14 (2): 34–9.
- Ernsberger P., Koletsky R.J. The Glucose Tolerance Test as a Laboratory Tool with Clinical Implications. INTECH. 2012: 3–14.
- Kastin A.J., Pan W., Maness L.M. Koletsky R.J., Ernsberger P. Decreased transport of leptin across the blood-brain barrier in rats lacking the short form of the leptin receptor. Peptides. 1999. Vol. 20: 1449–53.
- Turley SD & Hansen CT (1986) Rates of sterol synthesis in the liver and extrahepatic tissues of the SHR/N-corpulent rat, an animal with hyperlipidemia and insulin-independent diabetes. Journal of lipid research. 1986. Vol. 27: 486–96.
- Hariya N., Mochizuki K., Inoue S., Morioka K., Shimada M., Okuda T., Goda T. Insulin Resistance in SHR/NDmc-cp Rats Correlates with Enlarged Perivascular Adipocytes and Endothelial Cell Dysfunction in Skeletal Muscle. Journal of Nutritional Science and Vitaminology. 2014. Vol. 60 (1): 52–9.
- Charles River. Research model. Электронный ресурс [https://www.criver.com
- Tong Y.C., Wang C.J., Cheng J.T. The role of nitric oxide in the control of plasma glucose concentration in spontaneously hypertensive rats. Neuroscience Letters. 1997. Vol. 233 (2–3): 93–6.
- Kurtz T.W., Morris R.C. Biological Variability in Wistar-Kyoto Rats Implications for Research with the Spontaneously Hypertensive Rat. Hypertension. 1987. Vol. 10 (1): 127–31.
- Ingalls A.M., Dickie M.M., Snell G.D. (1950) Obese, a new mutation in the house mouse. Journal of Heredity. 1950. Vol. 41: 317–8.
- Katsuda Y., Ohta T., Shinohara M., Bin T., Yamada T. Diabetic mouse models. Open Journal of Animal Sciences. 2013. Vol. 3 (4): 334–42.
- Ioffe E., Moon B., Connolly E., Friedman J.M. Abnormal regulation of the leptin gene in the pathogenesis of obesity. Proceedings of the National Academy of Sciences. 1998. Vol. 95: 11852–7.
- Panchal S. K., Brown L. Rodent Models for Metabolic Syndrome Research. Journal of Biomedicine and Biotechnology. 2011. Article ID 351982. DOI:10.1155/2011/351982.
- Jung U.J., Lee M.-K., Park Y.B., Jeon S.-M., Choi M.-S. Antihyperglycemic and antioxidant properties of caffeic acid in db/db mice. Journal of pharmacology and experimental therapeutics august. 2006. – Vol. 318 (2): 476–83.
- Kennedy A. J., Ellacott K.L., King V. L., Hasty A. H. Mouse models of the metabolic syndrome. Disease Models and Mechanisms. 2010. Vol. 3 (4): 156–66.
- Ste Marie L., Miura G.I., Marsh D.J., Yagaloff K., Palmiter R.D. A metabolic defect promotes obesity in mice lacking melanocortin-4 receptors. Proceedings of the National Academy of Sciences. 2000. Vol. 97: 12339–44.
- Tschop M., Heiman M.L. Rodent obesity models: an overview. Experimental and Clinical Endocrinology and Diabetes. 2001. Vol. 109: 307–19.
- Warbritton A., Gill A.M., Yen T.T., Bucci T., Wolff G. L. Pancreatic islet cells in preobese yellow Avy/- mice: relation to adult hyperinsulinemia and obesity. Proceedings of the Society for Experimental Biology and Medicine. 1994. Vol. 206: 145–51.
- Chakraborty G., Thumpayil S., Lafontant D.E. Woubneh W., Toney J.H. Age dependence of glucose tolerance in adult KK-Ay mice, a model of non-insulin dependent diabetes mellitus. Lab Animal. 2009. Vol. 38: 364–8.
- Taketomi S., Tsuda M., Matsuo T., Iwatsuka H. and Suzuoki Z. Alternations of hepatic enzyme active- ties in KK and yellow KK mice with various diabetic states. Hormone and Metabolic Research. 1973. Vol. 5: 333–9.
- Hofman C., Lorenz K. and Colca J.R. Glucose transport deficiency in diabetic animals is corrected by treatment with oral antihyperglycemic agent pioglitazone. Endocrinology. 1991. Vol. 129: 1915–25.
- Koranyi L., James D., Mueckler M., Permutt M.A. Glucose Transporter Levels in Spontaneously Obese (db/db) Insulin-resistant Mice. European Journal of Clinical Investigation. 1990. Vol. 85: 962–7
- Kuklin A.I., Mynatt R.L., Klebig M.L., Kiefer L.L., Wilkison W.O., Woychik R.P., Michaud E.J. Liver-specific expression of the Agouti gene in transgenic mice promotes liver carcinogenesis in the absence of obesity and diabetes. Molecular Cancer. 2004. Vol. 3 (17): 1–10.
- The Jackson Laboratory. Strain datasheet. Электронный ресурс [https://www.jax.org]
Поиск



